含挥发油饮片草豆蔻标准汤剂的质量评价方法研究
2023-11-06吴远波张鸿刘宇政焦涛冷红文吴克勤吴靓
吴远波,张鸿,刘宇政,焦涛,冷红文,吴克勤,吴靓
(1.江西省医学科学院,江西南昌,330006;2.丹东市市场监管事务服务中心药检部,辽宁丹东,118002)
0 引言
2021年2月,国家药品监督管理局在《中药配方颗粒质量控制与标准制定技术要求》( 以下简称《技术要求》)[1]中,首次以官方文件的形式提出了标准汤剂的概念:“标准汤剂系遵循中医药理论,按照临床汤剂煎煮方法规范化煎煮,固液分离,经适当浓缩制得并经适宜方法干燥制得,为衡量单味中药配方颗粒是否与其相对应的单味中药饮片临床汤剂基本一致的物质基准。”在中药配方颗粒的研制过程中,标准汤剂起到了重要的桥连作用,中药配方颗粒的所有药学研究均须与标准汤剂进行对比。
草豆蔻为姜科植物草豆蔻(Alpinia katsumadaiHayata)的干燥近成熟种子[2,3],具有燥湿行气、温中止呕的功效,用于寒湿内阻、脘腹胀满冷痛、暧气呕逆、不思饮食[2]。草豆蔻主要含有挥发油、黄酮、二苯庚烷等[4,5],具有保护胃黏膜、抗胃溃疡、促胃肠动力、镇吐、抗炎、抗肿瘤、抗氧化等药理作用[5]。本研究选用主产区且符合药典要求的10批草豆蔻饮片制备标准汤剂,以山姜素、乔松素为指标成分,并进行含量测定,计算两成分的含量转移率[6],测定各批次标准汤剂挥发油含量并计算其转移率,测定出膏率,并以标准汤剂进行HPLC指纹图谱研究,为草豆蔻标准汤剂作为物质基准的应用奠定基础。
1 实验部分
1.1 试剂和仪器
1.1.1 仪器
Thermo Scientific UltiMate 3000高效液相色谱仪(Thermo Fisher 公司,包括四元梯度泵,WPS-3000SL自动进样器,TCC-3000RS柱温箱,DAD-3000检测器);RE-52AA旋转蒸发器(上海亚荣生化仪器厂); EX125ZH电子天平(奥豪斯仪器有限公司,感量0.01mg);LE204E/02电子天平(梅特勒-托利多仪器〈上海〉有限公司,感量0.1mg);CP213电子天平(奥豪斯仪器有限公司,感量 1mg);SZCL-3A型数显智能控温磁力搅拌器(巩义市予华仪器有限公司);SHB-Ⅲ循环水式多用真空泵(郑州长城科工贸有限公司);KQ-500E超声波清洗器(昆山市超声仪器有限公司);DKM610C干燥箱(重庆雅马拓科技有限公司);DK-98-ⅡA电热恒温水浴锅(天津市泰斯特仪器有限公司)。
1.1.2 试剂与样品
试药:山姜素对照品(批号:PS010886,纯度大于98%)、乔松素(批号:PS010411,纯度大于98%)、小豆蔻明对照品(批号:PS000964,纯度大于98%)、桤木酮对照品(批号:PS010412,纯度大于98%)、草豆蔻对照药材(批号:PS030099),均购自成都普思生物科技股份有限公司。乙腈和甲醇为色谱纯,水为怡宝纯净水,甲醇及无水乙醇等其他试剂均为分析纯。
样品:10批不同来源草豆蔻饮片,经丹东市市场监管事务服务中心药检部焦涛主管中药师鉴定为草豆蔻(Alpinia katsumadai Hayata)的干燥近成熟种子,草豆蔻信息见表1。
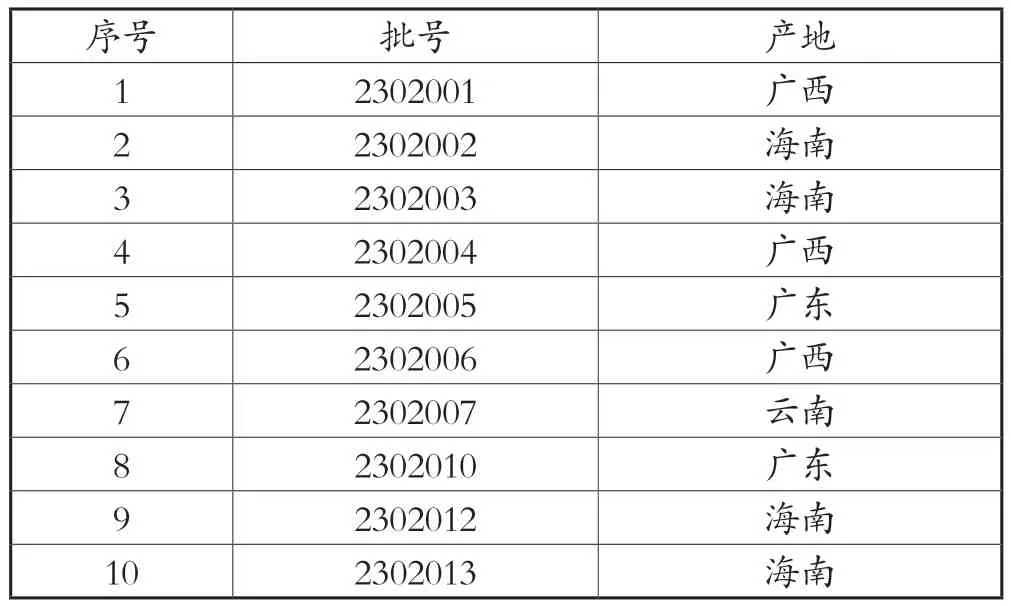
表1 10 批草豆蔻来源信息表
1.2 实验过程
1.2.1 草豆蔻饮片的检测
取草豆蔻饮片按《中国药典》2020年版一部“草豆蔻”饮片项下[2]标准进行检测,结果符合药典规定。
1.2.2 草豆蔻饮片标准汤剂的制备[7-9]
取草豆蔻饮片,捣碎,称取100.0g,加8倍量水浸泡30min,接挥发油提取器回流提取2h,接出挥发油,水提液趁热过滤,在药渣中再加6倍量水,接挥发油提取器回流提取30min,接出挥发油,水提液趁热过滤,合并两次滤液[10],减压浓缩至适量,将2次提取的挥发油合并,加入浓缩液中,定容至500mL,充分摇匀,得到规格为0.2g/mL的草豆蔻饮片标准汤剂。
1.2.3 草豆蔻标准汤剂指标成分含量测定
1.2.3.1 色谱条件
色谱柱为Agilent Zorbax SB C18柱(250mm×4.6mm,5μm);流动相为甲醇-水(60:40);检测波长为300nm;柱温为30℃;流速为1.0mL/min;进样量为5μL。该色谱条件下,供试品溶液各指标成分峰分离良好,见图1。
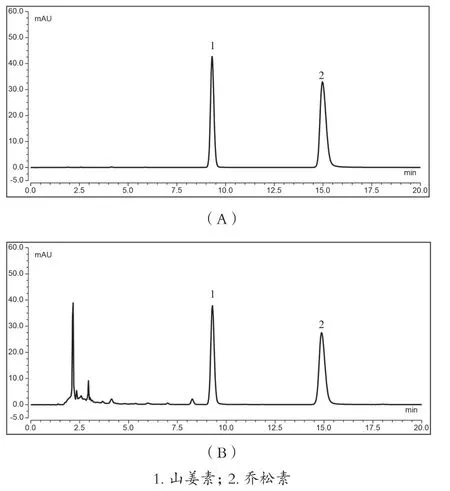
图1 山姜素对照品与乔松素对照品(A)、草豆蔻标准汤剂(B)HPLC图
1.2.3.2 对照品溶液的制备
取适量山姜素、乔松素对照品,精密称定,加甲醇制成每1mL含山姜素40μg、乔松素40μg的混合对照品溶液,即得。
1.2.3.3 供试品溶液的制备
取草豆蔻标准汤剂摇匀,精密量取1mL,置于10mL量瓶中,用无水乙醇稀释至接近刻度,超声处理(功率500W,频率40KHz)10min,冷却,用无水乙醇定容至刻度,摇匀,滤过,取续滤液,即得[11,12]。
1.2.3.4 线性关系考察
取适量山姜素、乔松素对照品,精密称定,加甲醇制成每1mL含山姜素100.0μg、乔松素105.6μg的混合对照品溶液。精密吸取该溶液配成系列浓度的对照品溶液,按“1.2.3.1”项下选定的条件测定,进样5μL,记录峰面积。以每个对照品的进样量(ng)为横坐标,峰面积为纵坐标,绘制标准曲线,得到2个成分的线性回归方程及线性范围,见表2。

表2 山姜素、乔松素的线性回归方程及线性范围
1.2.3.5 仪器精密度试验
精密吸取同一份对照品溶液5μL,按“1.2.3.1”项下选定的条件,连续进行6次的进样测定,记录2个指标成分的峰面积。山姜素、乔松素的峰面积的RSD分别为0.8%、0.9%。结果表明仪器精密度良好。
1.2.3.6 重复性
取同一批样品(批号:2302001),按“1.2.3.3”项下的方法制备供试品溶液6份,按“1.2.3.1”项下选定的条件进行测定,记录山姜素、乔松素的峰面积,计算2个指标成分的含量。结果显示,山姜素的平均含量为0.31mg/mL,RSD为1.0%;乔松素的平均含量为0.31mg/mL,RSD为0.9%。结果表明该含量测定方法的重复性良好。
1.2.3.7 稳定性试验
取草豆蔻标准汤剂同一份供试品溶液,分别在制备后0h、4h、8h、12h、24h,按“1.2.3.1”项下选定的条件进样5μL,记录2个指标成分的峰面积,考察样品溶液的稳定性。计算得山姜素、乔松素的峰面积的RSD分别为1.3%、1.4%。结果表明供试品溶液在24h内稳定。
1.2.3.8 加样回收率试验
取6份已知含量的样品(批号:2302001)摇匀,取6份,每份精密量取0.5mL,根据标准汤剂中山姜素、乔松素的含量按1:1的比例添加对照品[6](山姜素159.9μg、乔松素160.1μg),按“1.2.3.3”项下的方法制备,按“1.2.3.1”项下选定的条件测定,计算得山姜素平均回收率为103.6%,RSD为1.8%;乔松素平均回收率为101.1%,RSD为1.2%。结果表明本法具有良好的回收率。
1.2.3.9 样品测定
取10批草豆蔻标准汤剂样品,精密量取1mL,按“1.2.3.3”项下的方法制备,按“1.2.3.1”项下选定的条件测定,记录山姜素、乔松素的峰面积,计算10批标准汤剂中山姜素和乔松素的含量。结果见表3。
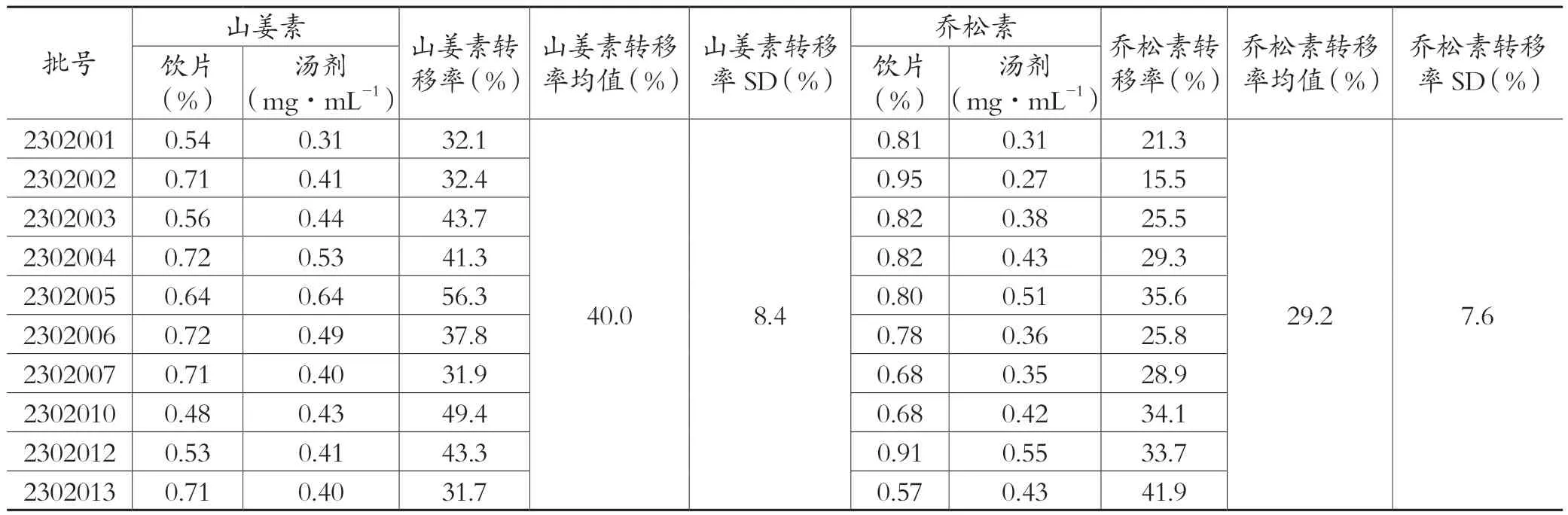
表3 草豆蔻标准汤剂山姜素、乔松素含量及转移率
1.2.4 指标成分及挥发油含量转移率和出膏率
根据标准汤剂中山姜素和乔松素的含量测定结果,计算两个指标成分的含量转移率,结果见表3。根据标准汤剂中挥发油含量测定结果,计算挥发油含量转移率,结果见表4。
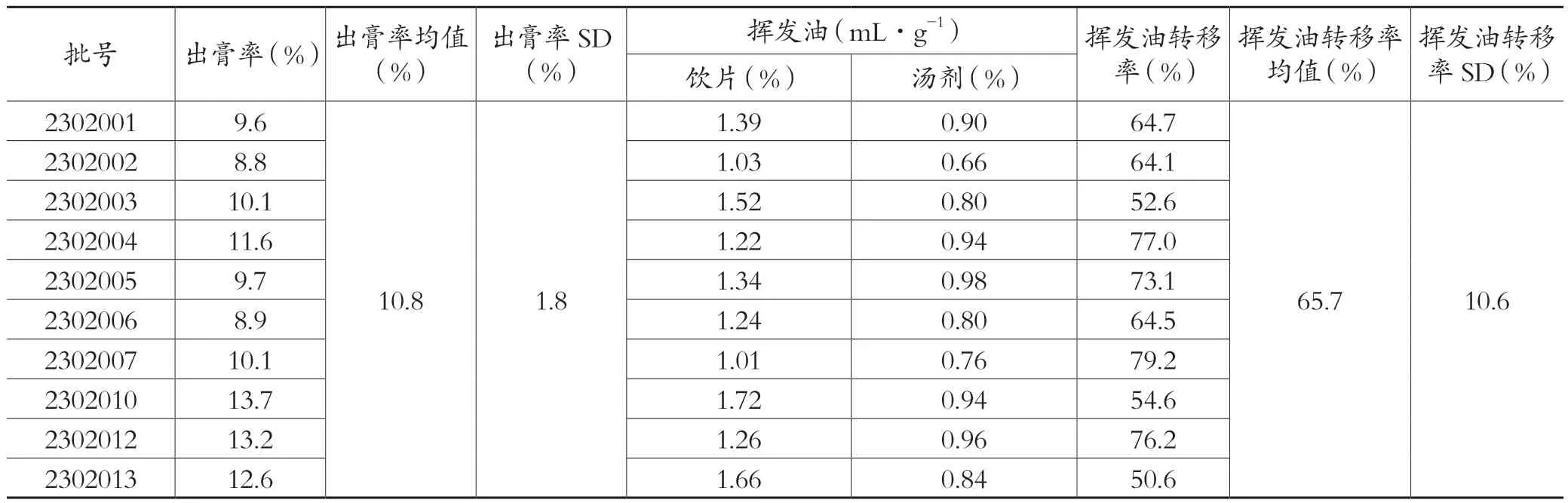
表4 草豆蔻标准汤剂出膏率及挥发油转移率测定结果
出膏率测定方法:取草豆蔻标准汤剂适量,摇匀,精密吸取10mL置于已干燥至恒重的蒸发皿中,水浴蒸干,105℃干燥3h,取出,置于干燥器中冷却30min,称定重量[13,14]。出膏率结果见表4。
1.2.5 草豆蔻标准汤剂HPLC指纹图谱测定
1.2.5.1 色谱条件
色谱柱为Agilent Zorbax SB C18柱(250mm×4.6mm,5μm);流动相为乙腈(A)-水(B),梯度洗脱(0~45min,40%~52%A;45~60min,52%~100%A);检测波长为262nm,柱温为40℃,流速为0.8mL/min;进样量为10μL。混合对照品与标准汤剂色谱图见图2。
经查阅文献[2,15],并比对对照品色谱图,判别1、2、6、8号峰分别为山姜素、乔松素、小豆蔻明和桤木酮。以保留时间适中、出峰稳定、有对照品的2号峰(乔松素)为参照峰(S),计算草豆蔻标准汤剂中主要共有峰的相对保留时间和相对峰面积[16]。
1.2.5.2 参照物溶液的制备
取山姜素、乔松素、小豆蔻明和桤木酮对照品适量,精密称定,加甲醇制成每1mL含山姜素40μg、乔松素40μg、小豆蔻明20μg、桤木酮20μg的混合溶液,即得对照品参照物溶液。取草豆蔻对照药材0.5g,置于具塞锥形瓶中,加甲醇50mL,超声处理(功率500W,频率40KHz)30min,放冷[17],摇匀,滤过,取续滤液,即得对照药材参照物溶液。
1.2.5.3 供试品溶液的制备
取“1.2.3.3”项制得的溶液,即得。
1.2.5.4 仪器精密度试验
精密吸取草豆蔻标准汤剂同一份供试品溶液10μL,按“1.2.5.1”项下选定的条件,连续进行6次进样测定,记录液相色谱图。以乔松素峰为参照,求得其余7个共有峰的相对保留时间及相对峰面积。结果显示,相对保留时间RSD<0.5%,相对峰面积RSD<5%,这表明精密度较好。
1.2.5.5 重复性试验
取同一批草豆蔻标准汤剂样品,按“1.2.3.3”项下方法平行制备6份供试品溶液,按“1.2.5.1”项下选定的条件进行测定。以乔松素峰为参照,求得其余7个共有峰的相对保留时间及相对峰面积。结果显示,相对保留时间RSD<0.5%,相对峰面积RSD<5%,这表明重复性良好。
1.2.5.6 稳定性试验
取草豆蔻标准汤剂(同一份供试品溶液),分别于制备后0h、4h、8h、12h、24h,按“1.2.5.1”项下选定的条件进样10μL。以乔松素峰为参照,求得其余7个共有峰的相对保留时间及相对峰面积。结果显示,相对保留时间RSD<0.5%,相对峰面积RSD<5%,这表明标准汤剂供试液在24h内稳定性良好。
1.2.5.7 样品测定与分析
取10批草豆蔻标准汤剂,按“1.2.3.3”项下方法分别制备供试品溶液,按“1.2.5.1”项下选定的条件进行测定,记录10批色谱图,用“中药色谱指纹图谱相似度评价系统(2012.130723版)”进行分析[18],生成对照指纹图谱,具体见图3、图4。10批标准汤剂指纹图谱与对照指纹图谱的相似度依次为0.978、0.934、0.969、0.987、0.984、0.988、0.989、0.991、0.930、0.920。草豆蔻标准汤剂指纹图谱中确定了8个共有峰,10批草豆蔻标准汤剂与对照图谱之间的相似度均>0.9[16]。
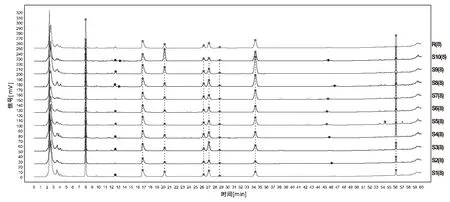
图3 10批草豆蔻饮片标准汤剂(S1~S10)的高效液相指纹图谱及对照指纹图谱(R)
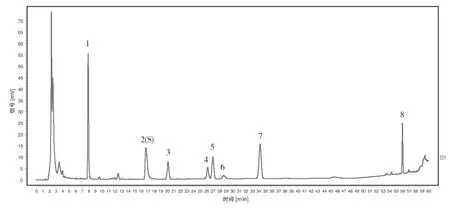
图4 草豆蔻标准汤剂对照指纹图谱
1.2.5.8 对照药材、标准汤剂指纹图谱比较
取对照药材参照物溶液,按“1.2.5.1”项下选定的条件进行测定,记录色谱图,见图5。结果显示,对照药材具有与标准汤剂相同的8个共有峰,表明两者化学成分无明显变化,具有一定的等效性与相关性[19]。
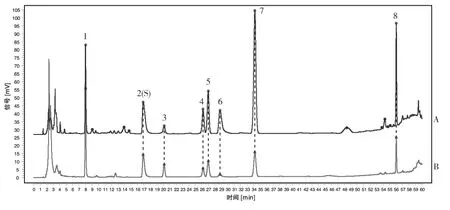
图5 草豆蔻对照药材指纹图谱(A)与标准汤剂对照指纹图谱(B)
2 结果与讨论
草豆蔻富含挥发油,为了更好地保留这类成分,制备标准汤剂时,需要将其单独提取出来,参考文献中的提取工艺[7-9],将提取得到的草豆蔻挥发油兑入浓缩的汤剂中。本研究所用的10批草豆蔻饮片的挥发油含量为1.01%~1.72%(mL/g),草豆蔻标准汤剂的挥发油含量为0.66%~0.98%(mL/g),挥发油含量转移率为50.6%~79.2%。数据显示,草豆蔻标准汤剂挥发油转移率较高。
草豆蔻饮片标准汤剂是以水为溶媒经标准化工艺提取的,其提取物大部分为水溶性物质。在《中国药典》2020年版草豆蔻饮片[2]的4个含量测定指标成分中,我们选取了水溶性相对高的山姜素和乔松素作为标准汤剂的指标成分,考察其含量转移率。而小豆蔻明和桤木酮的水溶性较差,含量转移率较低,我们将该两种成分放在指纹图谱测定中加以监控。
在指纹图谱分析方法建立过程中,使用DAD检测器在190~400nm全紫外波长扫描样品光谱图,提取254nm、262nm、280nm、300nm处的色谱图进行比对,并参考相关文献[15],综合考虑色谱峰数、色谱信号、分离情况、基线平稳情况等方面因素,最终选择262nm作为检测波长,同时,考察30℃、35℃、40℃柱温对色谱分离的影响。结果显示,柱温对色谱分离效果有较大影响,40℃时所得指纹图谱的分离效果较好,故建议柱温设定为40℃。
3 结论
本研究选取10批合格的草豆蔻作为研究用样品,建立了质量评价方法,得出山姜素的含量转移率范围为31.7%~56.3%,乔松素的含量转移率范围为15.5%~35.6%,挥发油含量转移率范围为50.6%~79.2%,出膏率范围为8.8%~13.7%,均符合《技术要求》的范围要求;建立了HPLC指纹图谱,标定了8个共有峰,指认了山姜素、乔松素、小豆蔻明和桤木酮峰,各标准汤剂图谱与对照指纹图谱的相似度均大于0.9。本文研究的标准汤剂出膏率、指标成分和挥发油含量转移率、指纹图谱三个参数是其作为物质基准的重要依据。本文的研究结果可以为草豆蔻配方颗粒后续的工艺研究、质量控制方法制定等提供参考[6, 20]。
