HPLC和UPLC-MS/MS法测不同水体中四环素类抗生素残留的比较研究
2023-11-06蔡鹏盛天露刘华陈冠宜易永刘永林王丽阳陈刚
蔡鹏,盛天露,刘华,陈冠宜,易永,刘永林,王丽阳,陈刚
(1.江西省检验检测认证总院检测认证技术发展研究院,江西南昌,330052;2.江西中医药大学药学院,江西南昌,330004;3.南昌医学院,江西南昌,330004)
关键字:HPLC;UPLC-MS/MS;四环素;比较研究
0 引言
四环素类抗生素是一类有并四苯结构的广谱抗生素,代表药物有土霉素(oxytetra-cycline,OTC)、四环素(tetracycline,TC)、金霉素 (chlortetracycline,CTC)、多西环素 (doxycycline,DC)等[1],当前,主要用于治疗革兰阳性细菌、革兰阴性细菌、支原体和立克次氏体等引起的感染[2]。由于缺乏完善的兽药抗生素使用监控系统,目前我国存在四环素滥用的情况。四环素类药物在动物体内代谢缓慢且容易蓄积,容易产生生物毒性,人体过量摄入还会引发潜在的“三致”(致癌、致畸、致突变)作用[3]。目前,四环素类抗生素的主要检测方法有高效液相色谱法、微生物法和液相色谱串联质谱法[4-7]等,由于微生物法只能测定四环素类抗生素的总量,因此,目前常用于检测四环素类抗生素的方法主要为高效液相色谱法和高效液相色谱-串联质谱法[8,9]。高效液相色谱-串联质谱法具有灵敏度高、分析时间快、定性分析结果可靠等优点,而高效液相色谱法则具有成本低,分离能力强等特点。本研究同时采用HPLC和UPLC-MS/MS法对不同水体进行四环素类抗生素含量测定,参照《食品安全国家标准产品中土霉素、四环素、金霉素和多西环素残留量的测定》(GB 31656.11—2021)中的仪器条件,从方法线性范围、检出限、精密度,加标回收率等角度出发,比较两法的异同点,以期为四环素在水中的快速测定提供新的检测方法。
1 仪器与试剂
1.1 仪器
1260 InfinityⅡ高效液相色谱仪(Agilent,美国安捷伦科技公司),AX224ZH/E电子分析天平(美国奥豪斯仪器常州有限公司);多功能振荡器;多管离心机(北京仪器设备有限公司);移液枪;1000mL容量瓶;UPLC-MS/MS-80050(岛津企业管理〈中国〉有限公司)。
1.2 试剂与材料
乙腈,甲醇(色谱纯,德国默克股份两合公司);草酸(优级纯,天津市大茂化学试剂厂);乙酸铵(色谱纯,德国莫尔斯公司);柠檬酸(分析纯,西陇科学股份有限公司);十二水合磷酸氢二钠,乙二胺四乙酸二钠(国药集团化学试剂有限公司);甲酸(色谱纯,美国埃尔公司);实验用水(超纯色谱用水);待测水体为南昌市青山湖区野外随机获取。HLB萃取小柱(上海安谱璀世标准技术服务有限公司);四环素、土霉素、金霉素、多西环素标准品(上海安谱璀世标准技术服务有限公司)。
0.1mol/L NaH2PO4溶液:称取14.20g NaH2PO4,用纯水溶解,转移于1000mL容量瓶中定容,摇匀备用。
0.1mol/L柠檬酸:称取21.01g柠檬酸,用纯水溶解,转移于1000mL容量瓶中定容,摇匀备用。
Mcllvaine缓冲溶液:将1000mL 0.1mol/L柠檬酸溶液与 625mL 0.1mol/L NaH2PO4溶液混合,使pH值位于3.0~8.0。
0.01mol/L Na2EDTA-Mcllvaine缓冲溶液:称取6.05g Na2EDTA溶解于1625mL前配Mcllvaine缓冲溶液中,混匀后,装至容器中备用。
0.01mol/L草酸溶液:称取1.26g草酸,用纯水溶解,转移于1000mL容量瓶中定容,摇匀备用。
2 实验方法
2.1 样品前处理
采用高效液相色谱法检测时,取300mL左右水样静置后过滤,加入50mL 0.01mol/L Na2EDTA-Mcllvaine缓冲溶液,再通过200mg / 6mL HLB固相萃取小柱(固相萃取小柱预先用5mL甲醇、5mL水二次淋洗),样品过完后,再加入5mL水淋洗,真空抽干。用2mL(20+80)甲醇水溶液溶解定容,过0.2um有机滤膜于2mL进样瓶,待测。采用高效液相色谱-串联质谱法检测时则取0.5mL水样静置后过滤并加入5mL 0.01mol/L Na2EDTA-Mcllvaine缓冲溶液即可,其余净化步骤与高效液相色谱法一致。
2.2 测定
2.2.1 高效液相色谱法
色谱柱:Welch XB-C18(250mm×4.6mm,5um),柱温:30℃,进样量:50μL:检测波长:350nm,流速1.0mL/min,流动相:甲醇-乙腈-0.01mol/ L草酸溶液,洗脱梯度见表1。
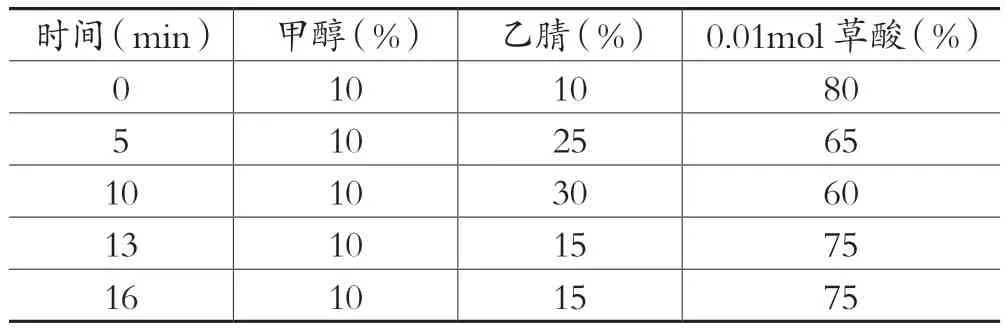
表1 WeIch XB-C18色谱柱洗脱梯度
2.2.2 高效液相色谱-串联质谱法
色谱柱:C18(2.1mm×150mm,5um),流动相:0.1%甲酸溶液-乙腈,流速为0.3mL/min,柱温为30℃,进样体积为5μL,洗脱梯度见表2。
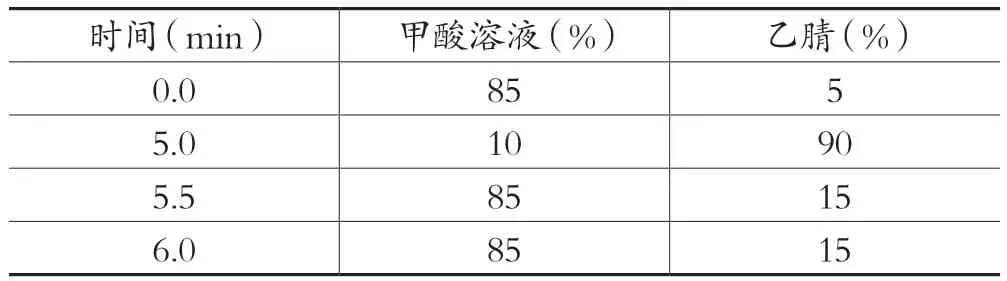
表2 C18色谱柱洗脱梯度
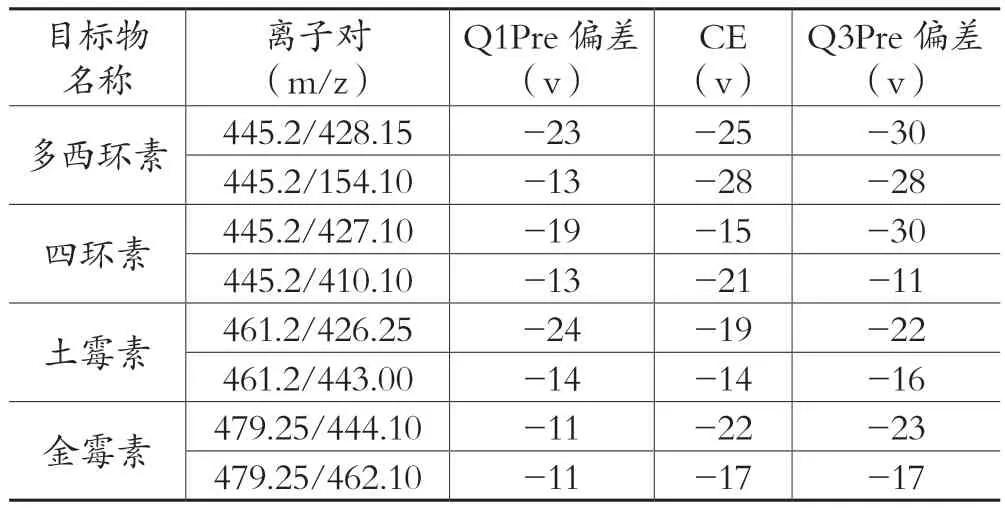
表3 质谱参数
质谱条件:离子源模式:电喷雾离子源(ESI+);检测方式:多反应监测 ( MRM);接口电压:4 kV;雾化器:氮气3L/min;干燥气;氮气10L/min;加热气;空气10L/min;DL管温度:250℃;加热块温度:400℃;接口温度:300℃,MRM具体参数详见表 3。
3 结果与讨论
3.1 线性方程
称取适量的四环素、土霉素、金霉素及多西环素标准品,加入甲醇定容,分别配制成浓度为5mg/L、10mg/L、15mg/L、20mg/L、50mg/L混标。按照高效液相色谱条件和方法进行分析,同时配置浓度为20μg/L、50μg/L、100μg/L、200μg/L、500μg/L混标,并按照高效液相色谱-串联质谱条件和方法进行分析,以标准溶液中各目标物浓度为横坐标,各四环素类目标物峰面积为纵坐标,绘制标准工作曲线,得到回归方程,结果显示两种方法的线性关系良好,具体如表4及表5所示

表4 高效液相色谱法线性关系

表5 高效液相色谱-串联质谱法线性关系
3.2 检出限
配置本实验预估方法检出限3~5倍浓度的样品,平行测定 7 次,对其结果的标准偏差值进行计算,按公式MDL = t( n-1,0.99)×S计算本方法的检出限。本研究高效液相色谱法的方法检出限结果如表6所示,高效液相色谱-串联质谱的方法检出限结果如表7所示。

图1 液相色谱图
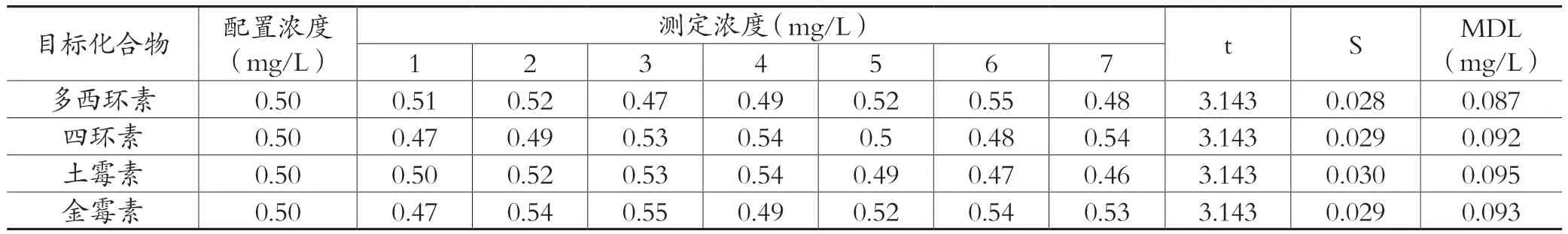
表6 高效液相色谱法的检出限结果
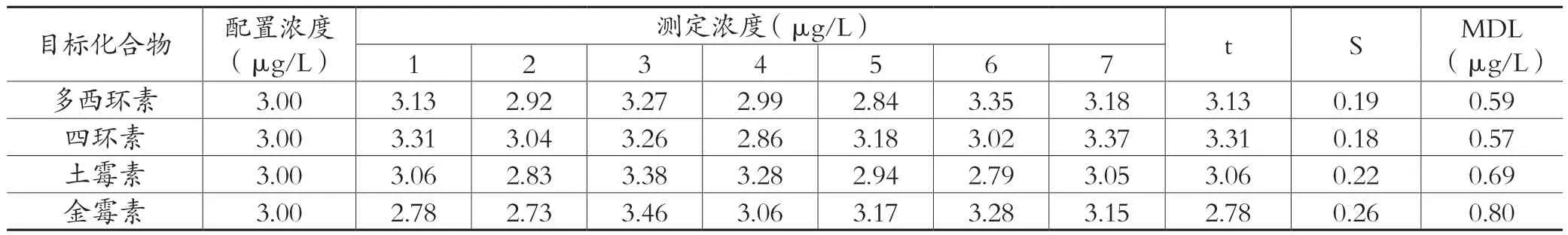
表7 高效液相色谱-串联质谱法的检出限结果
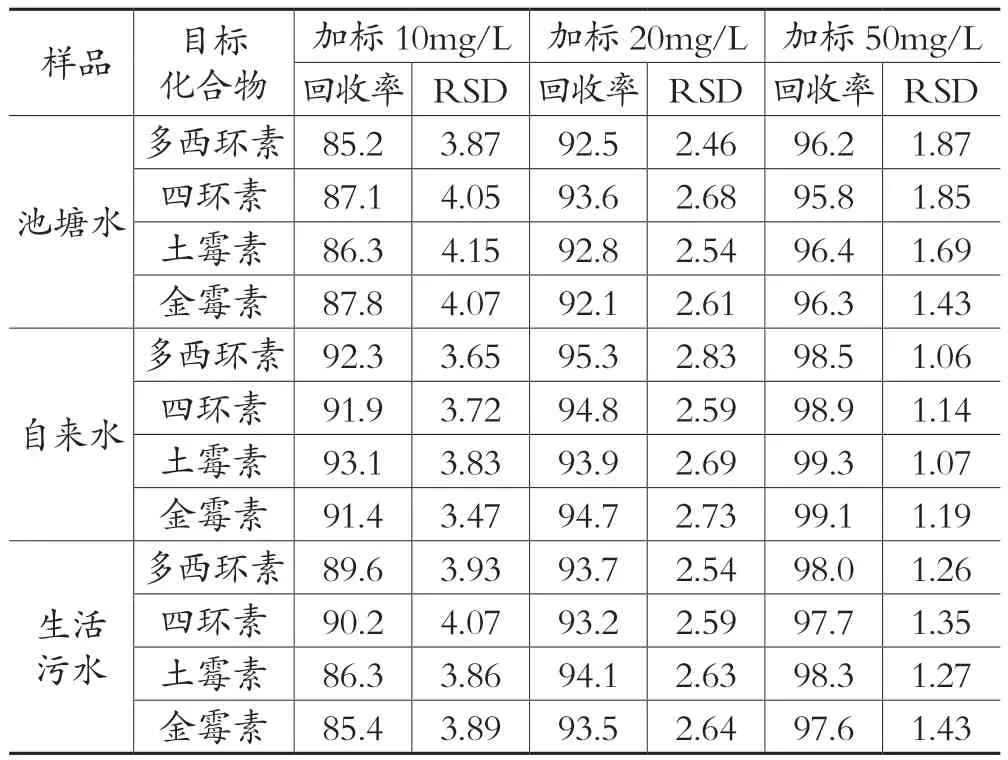
表8 高效液相色谱法的加标回收实验结果
3.3 加标回收率和精密度实验

图2 各化合物MRM谱图
分别选择适量的池塘水、自来水、生活污水,按照2.1的前处理方法处理,在2.2的色谱条件下进样分析,结果显示均未检测出多西环素、土霉素、四环素、金霉素。向3种水体样品中分别添加10mg/L、20mg/L、50 mg/L 、20μg/L、100μg/L、500μg/L 6个浓度水平的土霉素、四环素、金霉素混合标准溶液,其中10mg/L、20mg/L、50 mg/L浓度的加标样品采用高效液相法测定,20μg/L、100μg/L、500μg/L浓度的加标样品采用高效液相色谱-质谱法测定,重复6次并重新处理,高效液相法加标样品色谱图如图 1 所示,高效液相色谱-质谱法加标样品色谱图如图 2所示,计算测定结果的相对标准偏差和加标回收率。结果显示两种方法的加标回收率较高,详见表 8和表9。
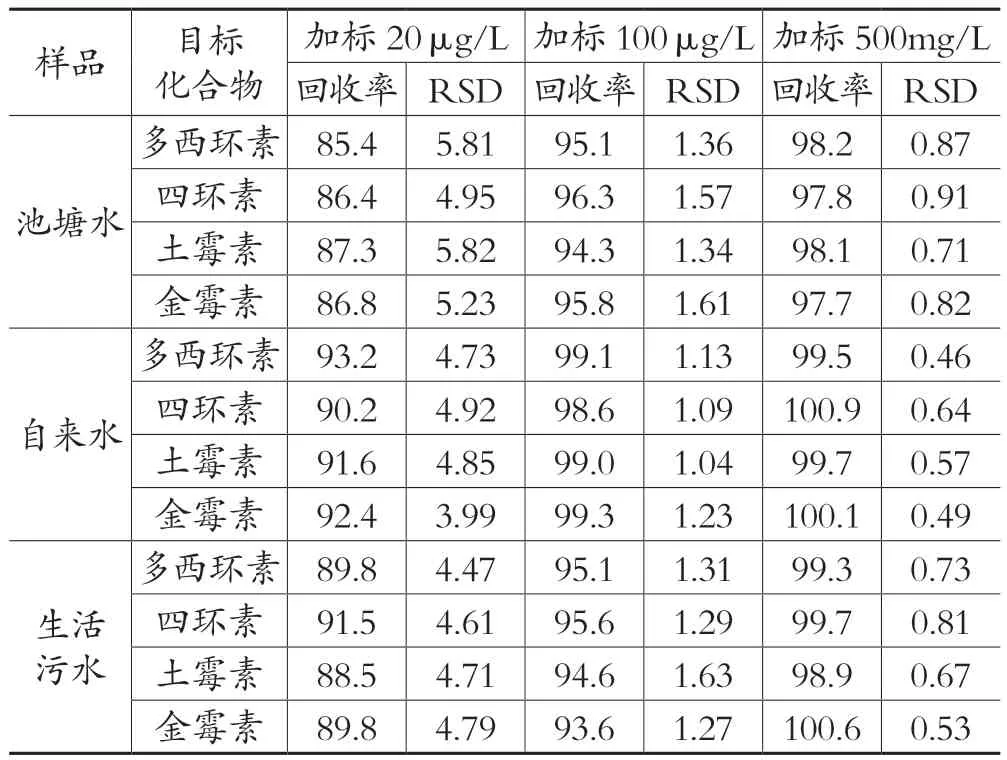
表9 高效液相色谱-质谱法的加标回收实验结果
4 总结
前期通过查阅文献,对目标物的前处理方法和仪器条件进行总结和参照,并对缓冲提取液浓度和体积、色谱条件选择等因素进行优化,确定了针对不同水体中四环素类残留量测定的最佳条件。在此条件下比较HPLC法和UPLC-MS/MS法的异同点,结果显示两种方法皆符合相关的检测方法要求,两种方法的线性关系良好,且回收率均在85.0%~100.9%,相对标准偏差均在0.46% ~4.15%,目标化合物在HPLC的检出限均低于0.1mg/L,在UPLC-MS/MS中的检出限均低于0.8μg/L。
