家族性VHL综合征患者家系突变类型及二次打击的分析
2023-09-27陈禹鑫逯艳文杨海洋张古田甘卫东郭宏骞
潘 骏,陈禹鑫,逯艳文,董 翔,杨海洋,张古田,甘卫东,郭宏骞
(1.南京大学医学院附属鼓楼医院泌尿外科,江苏南京 210008;2.江苏大学鼓楼临床医学院,江苏镇江 212000)
希佩尔·林道(Von Hippel Lindau,VHL)综合征是一种常染色体显性肿瘤,其特征是视网膜和中枢神经系统血管母细胞瘤、肾透明细胞癌(clear cell renal cell carcinomas,ccRCC)、嗜铬细胞瘤(pheochromocytoma,PCC)、胰岛肿瘤和内淋巴囊肿瘤[1]。VHL基因突变是VHL综合征的启动因素[2],根据突变类型为体系还是胚系而分为散发性VHL突变型ccRCC和家族性VHL综合征[3];文献报道,家族性VHL综合征的发病机理符合二次打击理论[4],甲基化及杂合性缺失都是导致患者VHL失活的重要机制[5-6],其中甲基化属于沉默抑癌基因的一种机制[7],杂合性缺失(loss of heterozygosity,LOH)则在多阶段致癌过程中可导致肿瘤抑制基因等位基因的丢失[8]。另外,单核苷酸多态性(single nucleotide polymorphisms,SNP)在患者发病中也起到了重要作用[9]。
本研究收集到一个家族性VHL综合征患者的外周血及病理样本,并提取DNA进行高通量基因检测,研究该家族性VHL综合征患者的胚系突变类型,并通过UCSCXena数据库、甲基化特异性聚合酶链式反应法(methylation specific polymerase chain reaction,MSP)、微卫星稳定性检测分别针对LOH、甲基化、突变及SNP进行实验及验证,分析患者可能的第二次打击位点及相关致病因素。
1 资料与方法
1.1 一般资料患者(先证者)男性,52岁,因左侧腰痛2月余于南京大学医学院附属鼓楼医院就诊,发现左肾肿瘤,遂收住入院行左肾肿瘤剜除术。患者2010年于当地医院行右肾肿瘤剜除术,术后病理提示ccRCC。患者分别于2013年10月、2014年2月于当地医院发现双侧肾上腺占位,并行双侧肾上腺切除术,术后病理提示PCC。2016年因右肾肿瘤复发,于上海市长海医院行右肾切除术,术后病理提示右肾ccRCC。
经过患者本人及家属同意后,对其家系中22人进行研究,其中8人患有多系统疾病,2人曾被考虑为VHL综合征但并未行进一步病理诊断(图1)。患病者分别为:Ⅰ1(先证者的母亲)患肾肿瘤、Ⅱ8(先证者)患双侧肾ccRCC合并双侧肾上腺PCC、Ⅱ2(先证者的哥哥)患单侧肾肿瘤、Ⅱ4(先证者的哥哥)患视网膜血管母细胞瘤、Ⅱ9(先证者的姐姐)患单侧肾肿瘤、Ⅲ1(先证者的侄女)患双侧肾上腺PCC、Ⅲ2(先证者的侄女)患颅内血管肿瘤、Ⅲ7(先证者的侄子)患双侧肾ccRCC合并双侧肾上腺PCC。
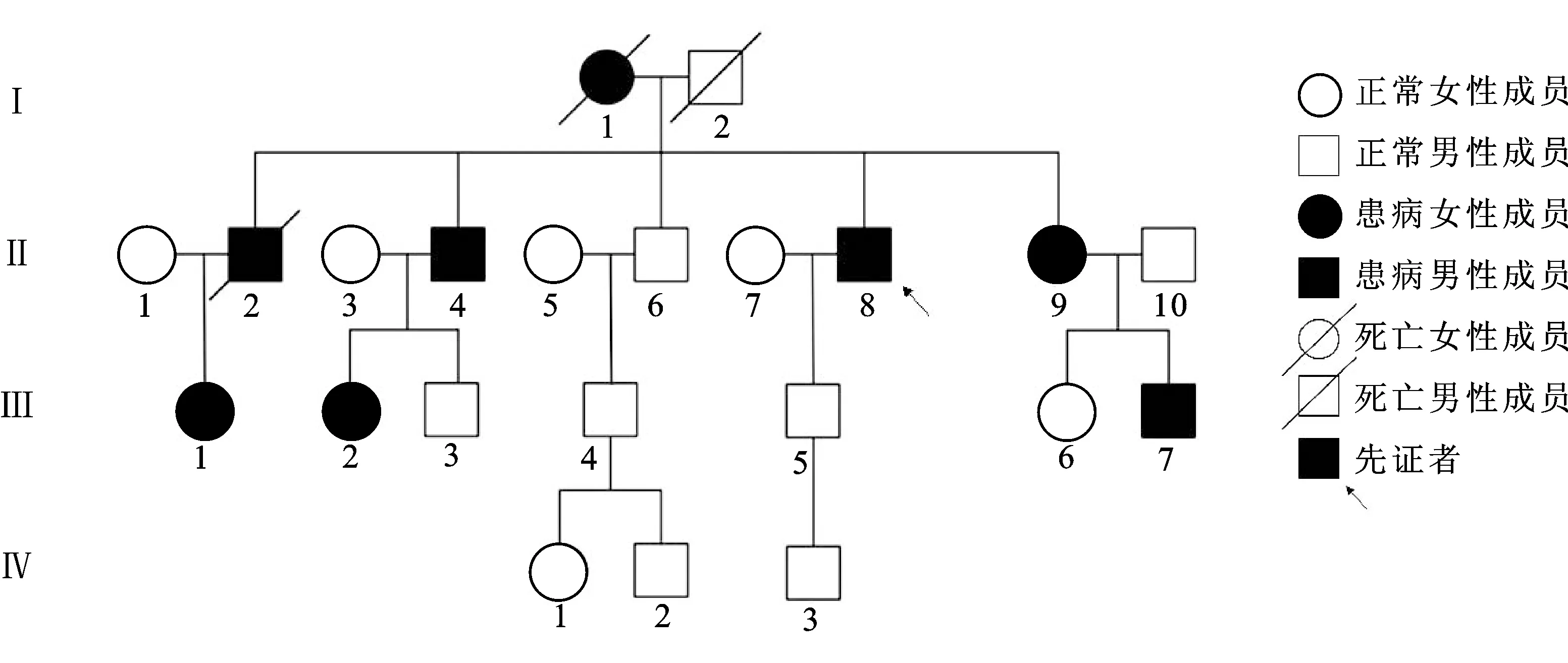
图1 VHL综合征患者家族系谱图
1.2 试验方法
1.2.1高通量测序(next generation sequencing,NGS)[10]将患者肾肿瘤切除术后的病理白片送上海仁东医学科技有限公司测序后进行序列分析,使用基于单分子聚合酶链式反应(single molecule-polymerase chain reaction,SM-PCR)技术的肿瘤基因突变检测试剂盒富集血液样本关于遗传性肿瘤等642个相关基因区域的 ctDNA 片段,使用NextSeq CN500基因测序仪,对相关基因的全部外显子区域进行捕获测序,在DNA水平上分析相关基因的点突变、插入缺失、重排、拷贝数等突变形式。
1.2.2甲基化位点预测研究 UCSCXena数据库(http://xena.ucsc.edu/)是一个基因组相关数据库,汇集了约200个公共数据库,包括癌症基因组图谱计划(the cancer genome atlas,TCGA)数据库、癌细胞百科全书(cancer cell line encyclopedia,CCLE)数据库等。该数据库可用于检查拷贝数和甲基化、体细胞突变、基因表达、蛋白质表达和可视化,以及解释癌症基因组学数据可视化和解释癌症基因组学数据[11]。依据UCSCXena数据库中包含的TCGA数据库病例作为病例筛选的基本数据,后根据VHL基因来添加数据集,选择所需要的甲基化数据集,分别选择仅包含启动子的区域,含270 000个甲基化位点的模式,以及包含启动子和基因内部的区域,有450 000个位点的模式,最终获得致病的甲基化突变位点。
1.2.3MSP检测VHL基因甲基化[12]①采用200 μL新鲜血液提取基因组。②经亚硫酸盐转化进行甲基化修饰:取已提取的DNA 50 μL,加入2 mol/L NaOH溶液5 μL。37 ℃条件下变性10 min。加入10 mmol/L对苯二酚30 μL、3 mol/L NaHSO3溶液520 μL。50 ℃水浴16 h进行甲基化修饰。③引物设计:针对VHL的转录起始位点上游2 000 bp至下游100 bp是启动子区域,根据该区域设计甲基化与非甲基化的引物组。共设计出5组引物,每一组引物包括了上游引物及下游引物(MF:为甲基化的上游引物;MR:为甲基化的下游引物;UF:为非甲基化的上游引物;UR:为非甲基化的下游引物),同时从既往报道的VHL综合征甲基化病例中引用另一对引物[13]。④MSP:参考JAMES及参考文献等的方法[14],对转化的DNA进行PCR扩增,PCR反应条件:95 ℃热启动5 min,95 ℃变性30 s,56 ℃退火30 s,72 ℃延伸30 s共35个循环:用含溴化乙啶染色的1%琼脂以不同引物扩增的产物,凝胶成像自动分析系统观察分析结果[15]。

表1 引物序列表
1.2.4微卫星稳定性检测 患者行肾肿瘤切除术后其病理白片被送至上海仁东医学科技有限公司,进行测序及序列分析,微卫星-聚合酶链式反应(micro star international-polymerase chain reaction,MSI-PCR)技术的肿瘤基因微卫星位点检测试剂盒富集血液样本,使用NextSeq CN500基因测序仪,针对642个相关基因区域的全部外显子进行捕获。
1.2.5SNP位点检测 查阅参考文献,找到与VHL综合征相关的SNP位点,即rs1642742[16]、 rs779805[17],对所取样本进行全外显子捕获测序(whole exome sequence,WES),并与参考基因组进行比较,确定SNP的发生位点。
2 结 果
2.1 家系资料分析结果通过对先证者及其家系进行病史分析,能够发现该患者家系中的患者属于典型VHL综合征。根据既往研究可知,VHL综合征的初始症状可发生于全身多处器官,以合并PCC最为常见,多合并肾恶性肿瘤的发生,好发于20~40岁人群,男性多见。在患者家系中发现有3名家系成员罹患肾恶性肿瘤,1名家系成员罹患肾上腺PCC,同时有2名家系成员罹患肾恶性肿瘤合并双侧肾上腺PCC,且该患者家系成员发病年龄满足家族性VHL综合征早发的特点,十分符合VHL综合征的临床特征。
2.2 NGS高通量检测结果通过对患者基因点突变、插入缺失、重排、拷贝数等突变形式的分析,未发现明确的插入缺失、重排、拷贝数变化。存在2种胚系突变基因,分别是在第500位碱基处由G碱基变为A碱基的p.R167Q突变型VHL,位于第1 420位碱基的由C碱基变为T碱基的MUTYH,以及一种体系突变:位于第12 752位碱基的由C碱基变为T碱基的KMT2D。目前在已发现的突变中仅有VHL基因突变存在明确临床意义。
2.3 甲基化位点预测结果通过UCSCXena数据库验证ccRCC中VHL的DNA甲基化水平(图2A)。结果显示VHL的基因表达和DNA甲基化相关,可见3个与VHL相关的甲基化位点,数据库中包含二次打击位点非甲基化患者,因1号位点在所有数据库病例患者中均为高表达,遂予以排除。故致病甲基化位点分别为:cg20916523和cg25539131。为此我们根据甲基化数据库中所提供的2个甲基化位点进行MSP数据研究(图2B)。结果发现cg20916523在该患者中并未出现,cg25539131位于非CPG岛处的外显子处,而该位置中的甲基化意义并不明确,且一般并不影响基因的表达量[18],所以予以排除。同时,根据甲基化引物设计数据库设计5组引物行PCR研究,甲基化引物并未扩增出目的条带(图2C),所以在该VHL患者的基因上无甲基化证明,因此该患者第二次打击位点并非甲基化。
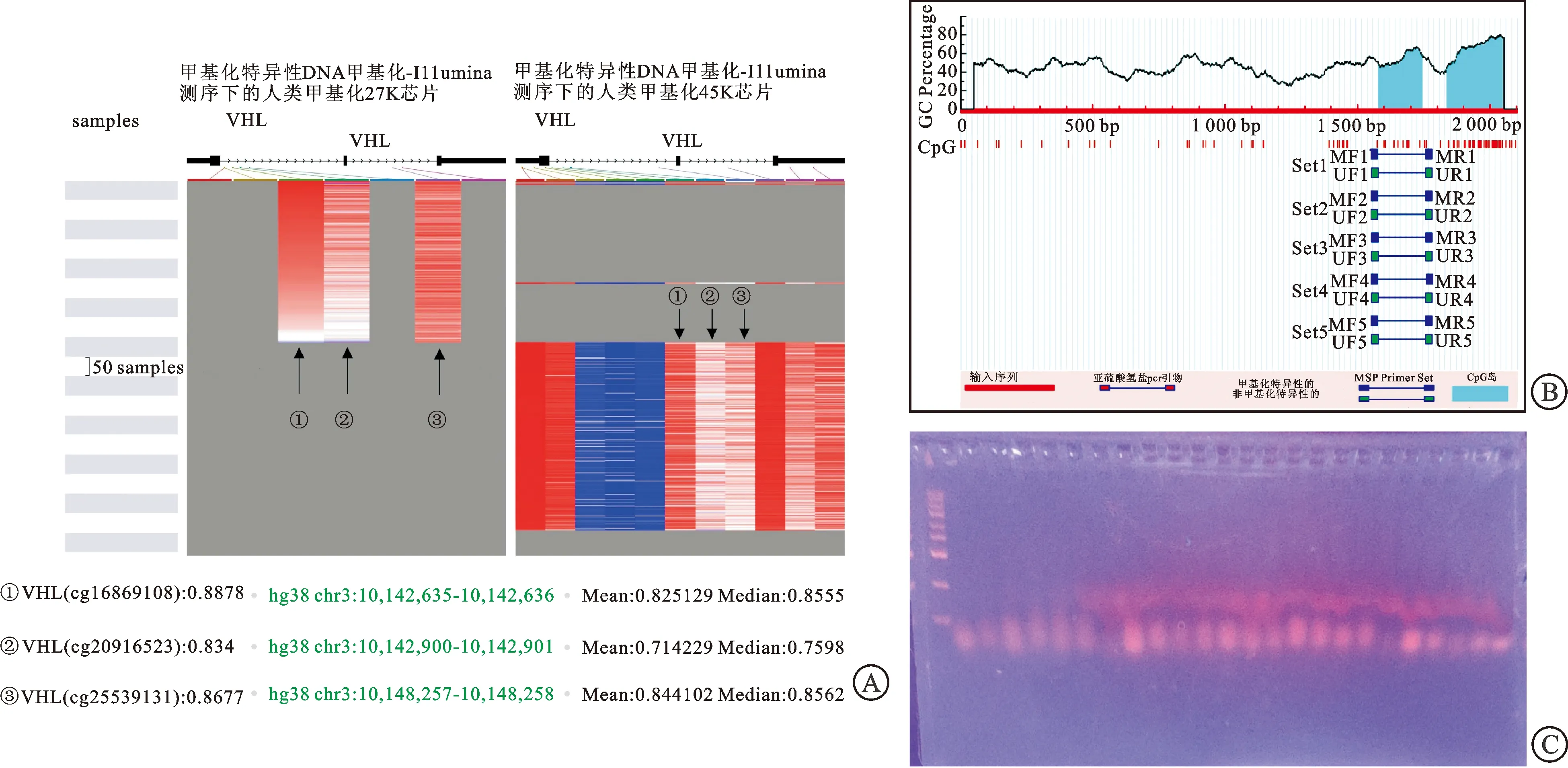
A:根据UCSCxena数据库结果,排除非VHL基因范围内的基因位点,留下3个位点,1号位点在所有样本中都高表达而排除,2号位点和3号位点为筛选出的甲基化位点;B:MSP中针对数据库提供的位点设立位点F1-5及R1-5,检测DNA组中甲基化的可能性;C:基因组荧光染色图,在染色图中未见明确甲基化位点。
2.4 SNP及杂合性缺失检测结果基于二代测序,我们通过MSI-PCR在VHL基因的相应片段中发现存在微卫星不稳定区域共91个,对此我们进行了微卫星状态分析,结果提示该患者微卫星稳定,可判断患者第二次打击位点并非LOH。依据WES结果,我们发现在rs779805处存在SNP,位于第183位核苷酸,密码子61 C/G。
3 讨 论
VHL综合征是一种罕见的家族遗传性疾病,能够累及全身多个脏器,临床表现异质性大[19-20]。根据表型特征,VHL综合征可分为4类:1型、2A型、2B型和2C型。1型中不存在PCC,2A型和2B型可能表现为PCC,并通过伴随的ccRCC额外风险进行区分,而2C型仅呈现ccRCC[21]。遗传性VHL疾病中最常见的突变R167Q属于2B型突变,发生此类突变易患ccRCC[22]。VHL综合征的致病基因即VHL基因为抑癌基因,定位于3号染色体短臂,含有3个外显子,编码pVHL蛋白质[23]。pVHL和缺氧诱导因子(hypoxia inducible factor,HIF)结合,引起HIF降解。当缺失或者变异导致pVHL合成减少时HIF积累,将可能引起下游基因持续激活而导致肿瘤的发生发展[24]。
根据Knudson的二次打击假设,通过对儿童视网膜母细胞瘤发病率的分析,认为该疾病的发生是二次突变的结果。家族型者第1次突变发生于生殖细胞,第2次突变发生于出生后体细胞,肿瘤的发生是由于肿瘤抑癌基因的双等位基因丢失,大量分子生物学研究已证实了这种由统计学分析提出的“二次打击”学说[25]。MAHER等[26]通过对收集到的VHL综合征患者进行相关性分析证实,VHL综合征的致病特点类似于视网膜母细胞瘤,符合二次打击学说,VHL疾病中的小脑血管母细胞瘤和肾细胞癌均发生在相同的突变模型,同时在肾细胞癌中第二次打击的原因以LOH、甲基化、基因突变等多见。
我们通过家系调查及对先证者外周血及病理组织采用NGS发现其为VHL综合征,VHL第3外显子发生病理性胚系错义突变,该突变类型为VHL综合征中最具特点的R167Q突变,目前已有通过下调HIF-2α来调节R167Q突变造成的缺氧、干扰可塑性调节的相关研究[22]。同时,我们针对第二次打击位点类型进行研究,采用数据库检索、MSP实验、微卫星稳定性研究及MSI-PCR等方法,发现该患者无甲基化、LOH、突变的情况,仅存在SNP。VHL突变的位点及类型众多,我们的研究不仅是对VHL综合征的进一步探索,同时也是对二次打击理论的进一步认识[27]。
SNP是导致患者肿瘤发生的重要机制之一,研究SNP能够对VHL综合征的发生发展有着更深刻的认识。目前对于SNP引起VHL综合征的机制尚不明确,但是既往研究认为SNP可能通过影响VHL基因的功能进而影响疾病易感性或者是通过促进LOH、甲基化的发生来产生作用,同时,疾病相关的SNP可能作为转录增强子,与转录因子相互作用,调节靶基因的表达[28],因此,对于SNP导致VHL综合征发生、发展的机制需要更进一步的研究与探讨。
VHL综合征有家族型和散发型之分,本研究中先证者患者确诊为VHL综合征,其发病的家系成员临床考虑为VHL综合征,且家族性恶性肿瘤病史伴有肾上腺恶性肿瘤疾病的家系成员数名,符合家族性VHL综合征诊断指征,因此均可临床确诊为VHL综合征[29],该家族也可初步判断为家族性VHL综合征。根据患者及其家系的发病年龄,发现患者与其家系其余患有肾癌者的发病年龄均早于40岁,符合家族性肾癌早发的临床特点[30],同时该患者家系中起初发病的位点并不一致以及易产生新的病灶,也符合该家族性肿瘤累及范围广、发展快的特点[31]。目前已知R167Q的突变均发生于胚系突变中,而R167Q突变导致VHL基因功能丧失,影响下游HIF-2特别是 HIF-2α 的组成性稳定,可能是促进其发展及多处转移的重要原因[22]。
综上所述,对于患者基因的不同层面,包括DNA、RNA、蛋白质层面的研究,有利于我们研究VHL综合征肿瘤的发生、发展机制。对于VHL综合征二次打击的研究能够对我们研究如何发现以及治疗提供了一个方向。同时,进行患者数据库基因甲基化研究对于帮助筛选患者甲基化等情况有着十分重要的作用,对符合二次打击学说的肿瘤有着很好的筛选作用。
