互为同分异构体的茚并咔唑类化合物的合成及性能研究
2023-08-30潘露露梁现丽陈婷温洁向陆军杭德余钱家盛
潘露露,梁现丽,陈婷,温洁,向陆军,杭德余,钱家盛
(1.安徽大学化学化工学院,安徽 合肥 230601;2.北京燕化集联光电技术有限公司,北京 102422)
有机发光二极管(OLED)在面世以来就以其优良的光电性质受到广泛关注,其具有成本低、质量小、光电响应速度快等优点,被誉为二十一世纪梦幻显示器[1-6]。而中间体的制备则对于其光电性质具有显著影响。咔唑是一种具有芳香性的稠环化合物,结构中氮元素含有孤电子对,且结构中存在刚性氮杂芳环,使其具有特殊的化学性质[7-9]。咔唑整体形成的共轭体系能够实现内部电子更好的转移,同时刚性结构能够让化合物本身具备更好的热稳定性和光学稳定性[10-12]。含有咔唑基团的材料或咔唑结构的衍生物在医药、染料、传感和光电等领域中得到广泛应用,特别是以咔唑为基础设计合成的光电材料,可在一定程度上使材料具有良好的光电性能[13-15]。
5,11-二氢-11,11-二甲基茚并[1,2-b]咔唑和11,12-二氢-12,12-二甲基茚并[1,2-a]咔唑互为同分异构体,它们都是合成有机发光材料的重要中间体。本文设计合成上述两种茚并咔唑化合物。首先,以联硼酸频哪醇酯和9,9-二甲基-2-溴芴通过超低温锂化反应生成9,9-二甲基芴-2-硼酸频哪醇酯,然后再与邻溴硝基苯经Suzuki偶联反应生成9,9-二甲基-2-(2-硝基苯基)-芴,最后经Cadogan 关环反应合成目标产物5,11-二氢-11,11-二甲基茚并[1,2-b]咔唑和11,12-二氢-12,12-二甲基茚并[1,2-a]咔唑(图1)。该合成方法原料廉价易得、操作简单、成本低廉,适合工业化生产。

图1 目标化合物的合成路线Fig.1 Synthetic route of the target compound
1 实验部分
1.1 主要仪器和试剂
核磁共振仪,1H NMR,Varian Unity 公司;液相色谱仪,SPD-20A型,岛津企业管理(中国)有限公司;液相色谱-质谱联用仪,LC-MS,LTQ Orbitrap XL/LTQ orbitrap XL;热重分析仪,TGA 5500 型;差示扫描量热仪,DSC-214,德国耐驰有限公司;电化学工作站,VERSASTAT3-200,普林斯顿。
9,9-二甲基-2-溴芴;联硼酸频哪醇酯;醋酸钾;[1,1'-双(二苯基膦基)二茂铁]二氯化钯;二氧六环;邻溴硝基苯;碳酸钾;四(三苯基膦)钯,分析纯,山西瑞科材料股份有限公司;四氢呋喃;亚磷酸三乙酯;邻二氯苯;所用试剂均为分析纯或化学纯,市售。
1.2 实验方法
1.2.1 化合物2的合成
向配有机械搅拌和温度计的5 L三口瓶中依次加入1.5 L 二氧六环,300 g(1.1 mol)9,9-二甲基-2-溴芴,306 g(1.21 mol)联硼酸频哪醇酯,222 g(2.27 mol)醋酸钾,3.9 g(0.005 mol)[1,1'-双(二苯基膦基)二茂铁]二氯化钯。开启搅拌至所有原料溶解,通入氮气进行保护,并逐步进行升温,使得物料在105℃~110℃回流反应12 h。待反应完全后,将反应液冷却至室温,抽滤得固体,滤饼用300 mL 乙酸乙酯淋洗,滤液过15 cm 硅胶柱,过柱液减压浓缩,得到450 g灰色油状物,加入450 mL乙醇,室温下搅拌12 h,析出固体,抽滤,并用100 mL 乙醇淋洗,烘干后得到类白色固体化合物2,m.p.:130℃~133℃,纯度99.5%,收率88.67%。1H NMR(400 MHz,CDCl3)δ:1.2(s,12H);1.69(s,6H);7.06(s,1H);7.16(m,2H);7.33(m,2H);7.90(m,2H)。13C NMR(CDCl3,150 MHZ),δ:24.7,30.9,45.9,88.1,121.6,123.2,126.7,128.4,129.6,133.1,134.8,141.0,147.8。MS(ESI)m/z:320.19 [M+H]+。
1.2.2 化合物3的合成
向配有机械搅拌和温度计的5 L 三口瓶中依次加入2.6 L 四氢呋喃、500 mL水、266 g(0.83 mol)化合物2、176 g(0.87 mol)邻溴硝基苯、343 g(2.49 mol)碳酸钾、9.5 g(0.008 mol)四(三苯基膦)钯,开启搅拌至所有原料溶解,通入氮气进行保护,并逐步进行升温,使得物料在65℃~70℃回流反应12 h。反应液冷却至室温,加入300 mL 水,用300 mL 乙酸乙酯萃取2次,萃取出的有机相过10 cm 硅胶柱,过柱液减压浓缩,得到260 g 黄色固体粗品。粗品用2 L乙醇70℃~75℃打浆1 h,降至室温后抽滤,40℃鼓风干燥箱烘6 h,得到232 g 黄色固体化合物3,m.p.:172℃~173℃,纯度99.8% ,收率88.5%。1H NMR(400 MHz,CDCl3)δ:1.69(s,6H);7.28(s,1H);7.38(s,1H);7.55(s,1H);7.75(m,2H);7.85(m,3H);8.01(s,2H);8.09(s,1H)。13C NMR(CDCl3,150 MHZ),δ:30.9,45.8,121.6,123.2,124.7,126.7,128.0,128.5,128.9,129.6,130.5,135.9,136.9,139.9,141.0,147.8。MS(ESI)m/z:315.13 [M+H]+。
1.2.3 化合物4-A和4-B的合成
向配有机械搅拌和温度计的2 L三口瓶中依次加入580 mL邻二氯苯、232 g(0.73 mol)化合物3、190 mL(1.1 mol)亚磷酸三乙酯,开启搅拌至所有原料溶解,通入氮气进行保护,并逐步进行升温,使物料在160℃~165℃回流反应2 h。将反应液冷却至45℃~50℃,油泵减压蒸出溶剂后得到棕色油状物。棕色油状物过20 cm硅胶柱,5 L[V(庚烷)∶V(乙酸乙酯)=40∶1]溶剂冲洗,过柱液减压浓缩,得到65 g类白色固体化合物4-A,m.p.257.58℃,纯度99%,收率31%。再用5 L[V(庚烷)∶V(乙酸乙酯)=10∶1]溶剂冲洗,过柱液减压浓缩,得到38 g类白色固体化合物4-B,m.p.:224.83℃,纯度99.19%,收率32.7%。4-A:1H NMR(400 MHz,CDCl3)δ: 8.12(m,2H);8.01(s,1H);7.75(m,1H);7.69(s,1H);7.50(m,1H);7.41(m,2H);7.36(d,1H,J=7.26 Hz);7.27(m,1H);1.62(s,6H)。13C NMR(CDCl3,150 MHZ),δ:30.9,40.7,111.1,116.8,119.4,121.6,122.6,123.2,124.4,126.7,127.6,129.6,141.2,147.8。MS(ESI)m/z:283.14 [M+H]+。4-B:1H NMR(400 MHz,CDCl3)δ:8.16(m,2H),8.13(s,1H),7.87(d,1H,J = 7.9 Hz),7.73(d,1H,J = 7.9 Hz),7.55(m,2H),7.48(m,1H),7.44(m,1H),7.39(m,1H),7.32(m,1H),1.76(s,6H)。13C NMR(CDCl3,150 MHZ),δ:30.9,45.8,97.8,110.4,119.8,121.4,123.2,126.7,127.9,129.6,133.5,135.1,144.0,147.8。MS(ESI)m/z:283.14 [M+H]+。
2 结果与讨论
2.1 通过核磁共振氢谱实现化合物4-A和4-B的区分
化合物4-A 和4-B 的同分异构仅仅表现在构造异构上,通过两者的核磁氢谱(图2、图3)不难看出,在α、β 两个碳上的氢核呈现截然不同的峰型。化合物4-A的αA和βA位置上的氢因为其周围并没有其他氢核影响,在氢谱上化学位移为δ=7.69 ppm 和δ=8.01 ppm 处表现为未发生偶合裂分的单峰,而化合物4-B的αB和βB位置上的氢由于相互影响,在氢谱上化学位移为δ=7.73 ppm 和δ=7.87 ppm 处表现为发生偶合而裂分的两个双重峰。通过核磁共振氢谱能够很好地对两者进行准确的区分,对两者的相关性能比对提供了支撑。
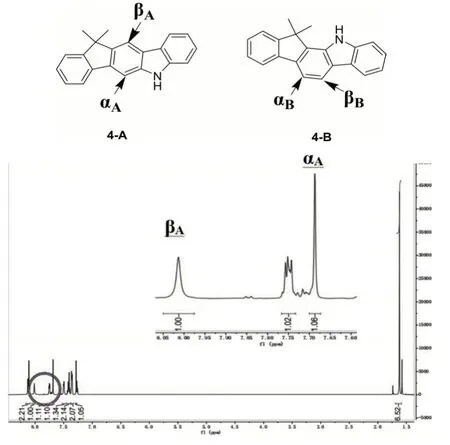
图2 化合物4-A的核磁共振氢谱图Fig.21H NMR spectrum of 4-A
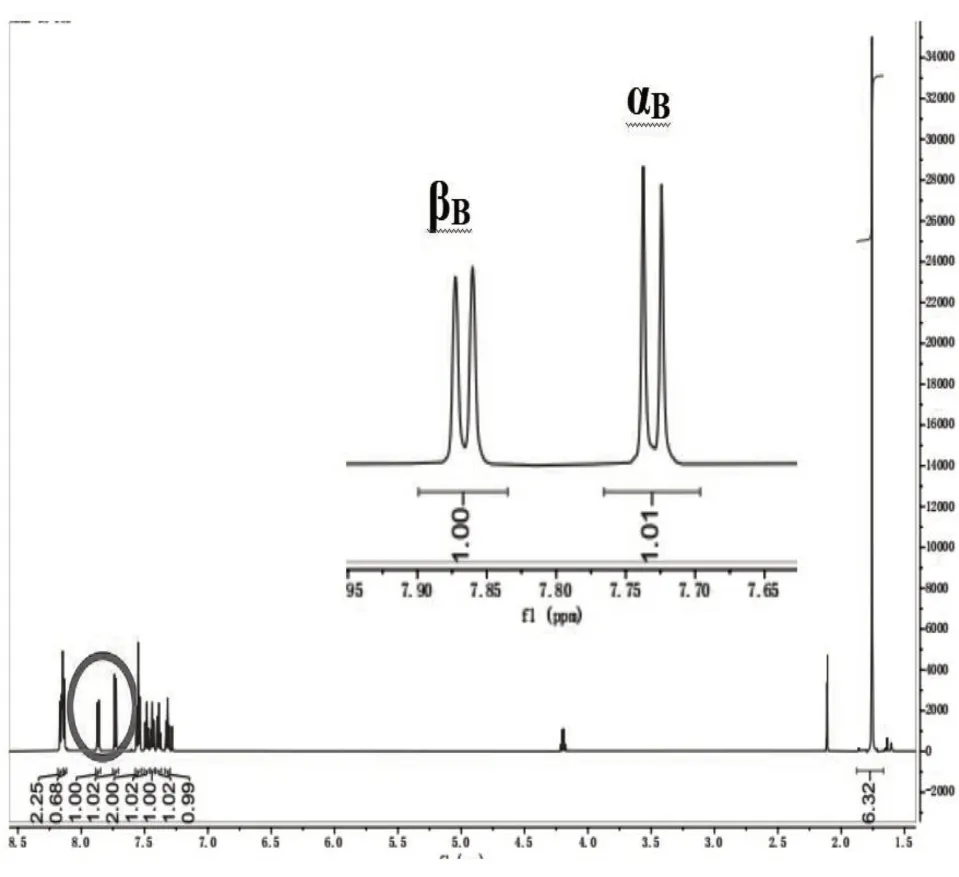
图3 化合物4-B的核磁共振氢谱图Fig.31H NMR spectrum of 4-B
2.2 化合物4-A和4-B的热力学性能研究
为研究化合物4-A 和4-B 的热力学性能,对它们进行TGA 和DSC 分析,以评估目标化合物的热稳定性。TGA 测试条件为从40℃开始升温,升温速率为20℃/min,最终升温至800℃,氮气气流速度为70 mL/min。DSC 的测试条件为从40℃开始升温,升温速率为10℃/min,所使用的氮气气流速度为50 mL/min。从图4 可以看出,化合物4-A 和4-B 的热分解温度(Td,失重1%时)分别出现在224.2℃及217.5℃,4-A 的Td高于4-B,较高的Td说明目标化合物具有较好的热稳定性。通过DSC 曲线(图5、图6)可以看出,4-A 和4-B 的熔点分别为257.58℃、224.83℃,分析是因为两者的关环位置不同导致。经过以上分析认为,这两种化合物具有良好的热力学性能和成为有机电致发光材料优异中间体的潜能。

图4 化合物4-A,4-B的TGA曲线Fig.4 TGA curves of compounds 4-A and 4-B

图5 化合物4-A的DSC曲线Fig.5 DSC curve of compound 4-A

图6 化合物4-B的DSC曲线Fig.6 DSC curve of compound 4-B
2.3 化合物4-A和4-B的密度泛函理论(DFT)计算
实验中采用DFT 选用基组为6-31G,杂化密度泛函为B3LYP的计算方法,以Gaussian09软件对分子结构以及前沿分子轨道(fronlier molecular orbital,FMO)的分布进行计算模拟[16]。通过DFT 计算得到的FMO 分布如图7 所示,4-A 和4-B 分子的HOMO 主要集中在咔唑基团上,LUMO 主要集中在芴基团上。理论计算中咔唑有较强的给电子能力,见表1。

表1 分子4-A和4-B的轨道能级计算结果Tab.1 Calculations of the orbital energy levels of molecules 4-A and 4-B
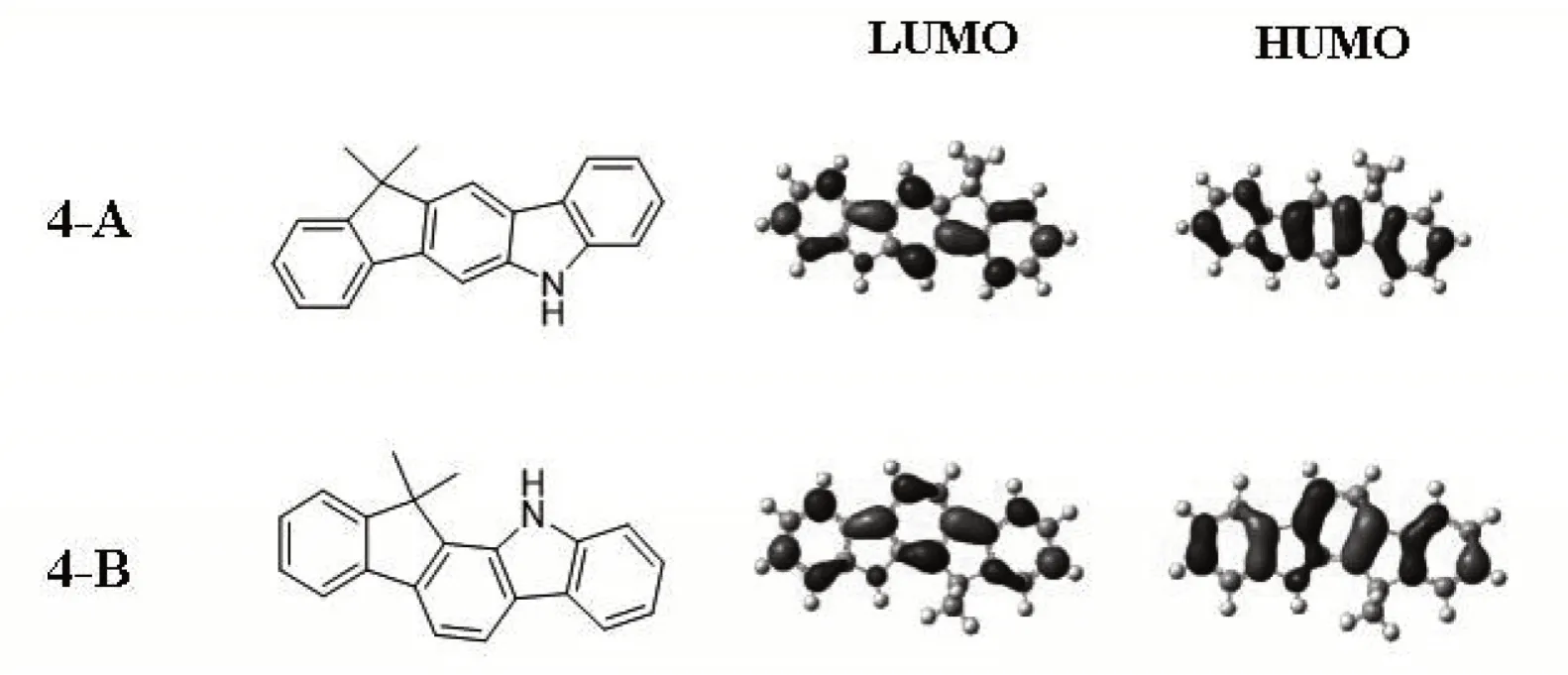
图7 分子4-A,4-B的HOMO、LUMO轨道分布Fig.7 HOMO,LUMO distribution of 4-A,4-B molecules
2.4 化合物4-A和4-B的电化学性能
通过循环伏安法,利用三电极测试体系(铂片电极为工作电极,Ag/AgCl 为参比电极,玻碳电极为对电极)对4-A 和4-B 的电化学性质进行研究。实验测得两种化合物的HOMO/LUMO 能级如表2 所示。相比于DFT理论计算的结果,实验测得的结果相对较小,不过和DFT 的数据结果具有相同的趋势。电化学测试结果表明,4-A 和4-B 具有较匹配的HOMO 和LUMO,使其具备开发为有机电致发光材料中间体的可能性。

表2 化合物4-A和4-B的电化学测试结果Tab.2 Compound 4-A and 4-B electrochemical test results
3 结论
本文以9,9-二甲基-2-溴芴和邻溴硝基苯为起始原料,经过三步反应同时制得两种化合物,再通过提纯工艺的改变使得两者分离得到高纯度的两种目标化合物,利用核磁氢谱分析将两种化合物加以区分。通过TGA、DSC 等发现两者的物性相似,具有较好的热稳定性,都能够作为良好的有机电致发光材料的中间体。通过模拟计算和实际测试相结合研究化合物轨道能级,为相似材料性能探索提供思路和方法。
