疾病状态下有机阴离子转运体表达和功能的变化及其机制研究进展
2023-08-22孙雪影刘李
孙雪影,刘李
(中国药科大学药学院药物代谢研究中心,江苏 南京 210009)
1 有机阴离子转运体概述
1.1 有机阴离子转运体结构
有机阴离子转运体(organic anion transporter,OAT)属于溶质转运体22(SLC22)亚家族。截至目前,研究发现SLC22亚家族OAT包括10个OAT(OAT1-10)和1个尿酸转运体(URAT1)。OAT约由540 ~ 560个氨基酸组成,共有12个跨膜结构域,游离的羧基和氨基定位在胞内侧。在第1个和第2个跨膜域之间有一个大的疏水环,环上有很多糖基化位点,而在胞内有一个含有多个磷酸化位点的疏水环[1]。人OAT与大鼠和小鼠OAT具有较高同源性,如人OAT1蛋白与其他哺乳动物OAT1同源基因具有86% ~ 96%的序列一致性;人OAT2蛋白与大鼠OAT2氨基酸序列同源性为79%;人OAT3蛋白与大鼠OAT3和小鼠OAT3的氨基酸序列同源性分别为79%和78%[2]。
1.2 有机阴离子转运体分布和功能特性
OAT的底物大致分为外源性药物及其代谢产物、内源性物质、环境毒素等。OAT系统最突出的特点是对底物的“多特异性”识别[3]。OAT1-3的底物具有较大的重合性。表1列举了OAT1-4的内源性底物和药物底物[4]。OAT介导主动转运过程,其中OAT1和OAT3属“三级主动转运”过程,即OAT1和OAT3通过与α-酮戊二酸交换方式逆负电荷转运有机阴离子,而α-酮戊二酸梯度的维持依赖于Na+-二羧酸协同转运,Na+梯度的维持依赖于Na+/K+-ATP酶[5]。而OAT7通过底物与短链脂肪酸交换介导有机阴离子的转运[6]。OAT8可能依赖于V-ATPase发挥转运功能[7]。OAT10可能是通过与琥珀酸交换方式逆负电荷实现有机阴离子的转运[8]。其他OAT亚型的转运机制尚不明确。
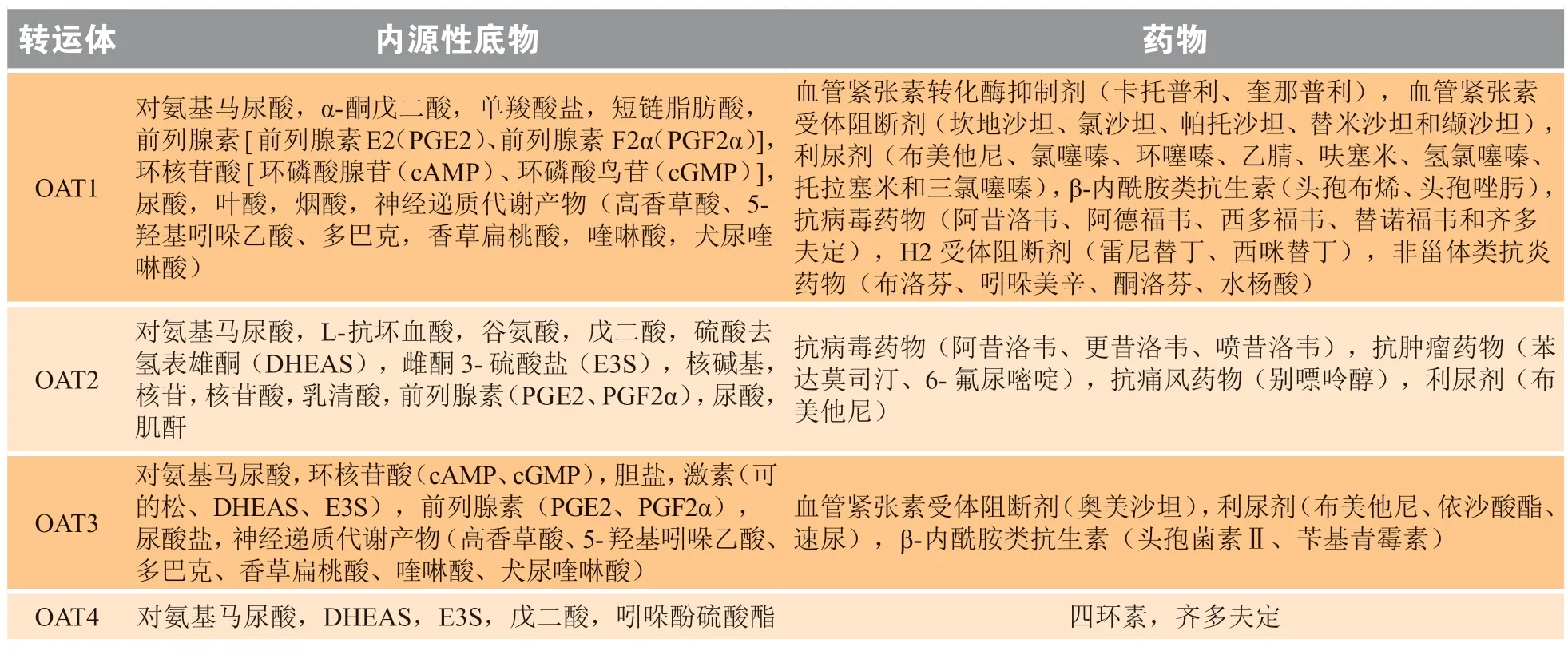
表1 有机阴离子转运体1-4的底物Table 1 Substrates of organic anion transporters 1-4
OAT的功能与分布是相对应的。OAT1最初由1997年Carlos E Lopez-Nieto等人鉴定并命名,其主要表达于肾脏,在大脑中有微弱表达[3]。OAT1定位在肾脏近端小管细胞基底侧,负责将血浆中有机阴离子转运到肾脏近端小管内皮细胞中,进一步由多药耐药相关蛋白外排至肾小管管腔[3]。OAT3最初从大鼠脑中分离得到。大鼠OAT3的mRNA在大鼠肾脏、脑及肝脏表达量较高,在眼中表达微弱。人肾脏OAT3的功能与OAT1一致。分布在脑微血管内皮细胞基底侧的OAT3介导有机阴离子从脑组织到血浆的转运,而分布在脉络膜细胞顶膜侧的OAT3可介导有机阴离子从脑脊液到血浆的转运[9-11]。OAT4和OAT5主要表达在肾脏近端小管细胞顶膜侧,OAT4介导尿酸等有机阴离子及全氟化学品从近端小管细胞到血液的重吸收[1,5]。OAT4在胎盘屏障上表达,定位在胎盘合胞体滋养细胞基底膜,介导激素、药物、毒素从胎儿侧到母侧的清除,对胎儿有保护作用[12]。OAT2最初是从大鼠肝脏中分离出来的一种肝脏特异性转运蛋白,在大鼠肝细胞基底侧表达水平较高,在肾脏中表达水平较低,在脑中不表达;OAT7最初在人体肝脏中被发现,主要分布在肝细胞基底侧。OAT2和OAT7负责将血浆中有机阴离子转运到胆汁中排出体外[13]。OAT2在小鼠中的表达存在性别差异,雄性小鼠的OAT2mRNA主要表达在肾脏,在肝脏微弱表达,而雌性小鼠的OAT2mRNA在肾脏和肝脏中表达量均较高,这可以解释药物和化学物质的药代动力学和毒性动力学的性别相关差异[14]。OAT6最初在小鼠嗅觉黏膜上皮细胞上被发现,另有研究表明其在睾丸中也有表达,在其他器官不表达。OAT8在大鼠肾脏集合管插层细胞顶膜侧中被发现,OAT9在小鼠肝脏和肾脏中被发现。OAT10表达在人肾脏近端小管细胞顶膜侧,在脑、心脏、小肠和结肠中也有表达,主要介导尼古丁和尿酸的转运[1]。URAT1最初在人肾脏中被发现,人和小鼠的URAT1在肾脏近端小管细胞顶膜侧高表达,主要介导尿酸的重吸收;小鼠脑毛细血管内皮细胞和脉络丛也表达URAT1[15]。表2总结了OAT在大鼠、小鼠和人体内的组织分布情况[1, 4, 16]。
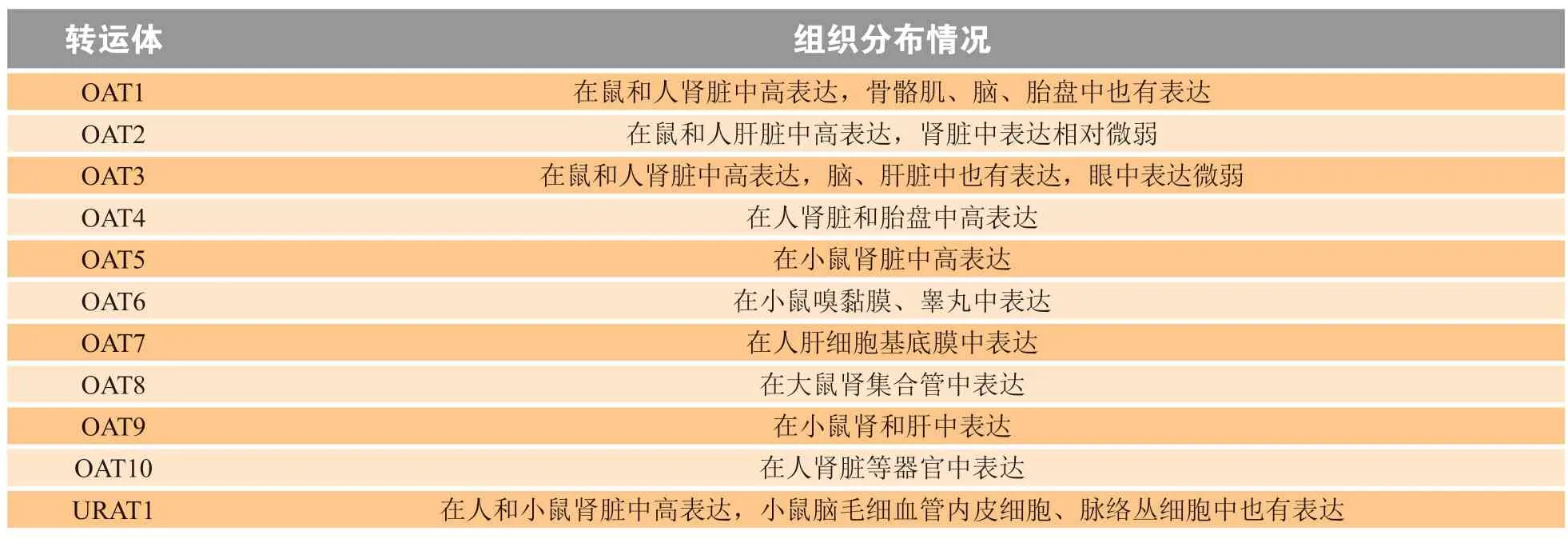
表2 有机阴离子转运体在大鼠、小鼠和人体内的组织分布Table 2 Tissue distribution of organic anion transporters in rats, mice and humans
OAT不仅对维持机体内环境稳态至关重要,而且参与多种临床药物的药物代谢动力学过程,如肾脏OAT1和OAT3通过影响抗病毒药、抗癌药、抗生素、抗高血压药、抗炎药等药物的排泄而影响药物的血药浓度和药效。血脑屏障上表达的OAT3是某些有机阴离子药物膜通透性较低的主要因素,如布美他尼通过作用于脑内神经元Na-K-Cl共转运体1(NKCC1)来治疗自闭症、癫痫和新生儿癫痫,但由于血脑屏障上OAT3的外排作用限制了脑内布美他尼的浓度,难以达到治疗效果。同时给予OAT3抑制剂丙磺舒可抑制血脑屏障上OAT3的功能,增加布美他尼的脑组织药物浓度,达到治疗脑疾病的效果[17]。
2 疾病状态下有机阴离子转运体的表达和功能变化及其机制
在某些病理情况下,OAT的异常变化可能会影响许多临床药物的治疗效果,阐明OAT在疾病状态下的表达和功能变化及其机制对深入了解疾病的发生发展及指导临床合理用药具有重要意义。表3总结了疾病状态下OAT的异常变化。
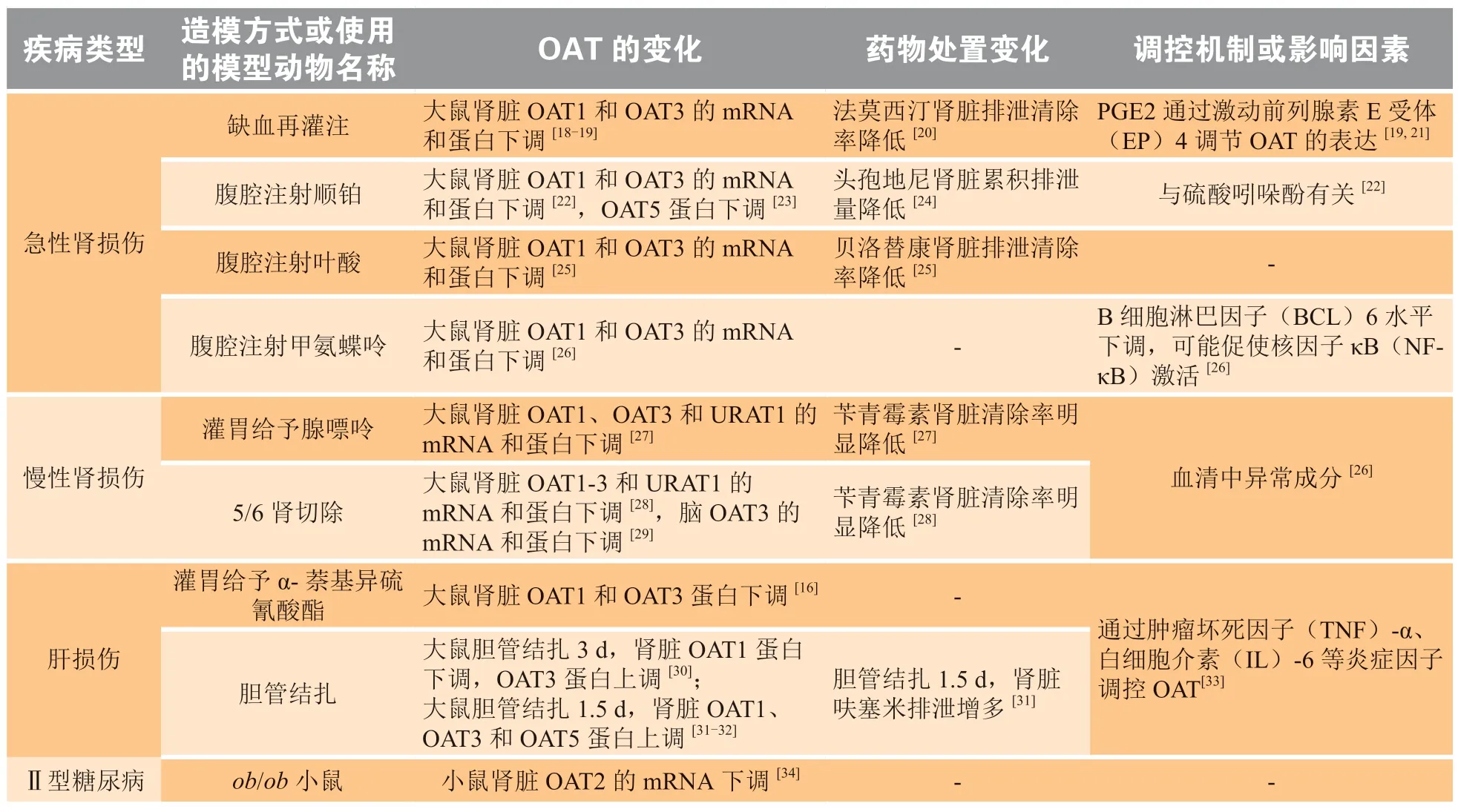
表3 疾病状态下有机阴离子转运体的表达和功能变化及其机制Table 3 Changes in expression and function of organic anion transporters and their mechanisms in disease states
2.1 肾损伤
2.1.1 急性肾损伤 急性肾功能衰竭是指短期内引起的双肾泌尿功能急剧障碍,以致机体内环境出现严重紊乱的病理过程。导致急性肾损伤的原因一般有2种——缺血再灌注和药源性急性肾损伤。肾毒性是很多临床治疗药物的毒副作用之一,如顺铂、庆大霉素、氨基糖苷类抗生素、环孢素、叶酸、甲氨蝶呤和造影剂等[46]。急性肾损伤会导致有机阴离子转运体的表达和功能下调,进而影响某些临床治疗药物以及内源性物质的消除。缺血再灌注诱导的急性肾损伤大鼠肾脏OAT1和OAT3的mRNA和蛋白表达水平均下调,文献表明这一过程是由环氧合酶(cyclooxygenase,COX)1代谢物通过EP4介导的。且有证据表明,低剂量的COX1抑制剂吲哚美辛和EP4拮抗剂L161982均可逆转这一过程[18-19]。另有体外实验表明,COX1代谢物PGE2可呈时间和浓度依赖性下调NRK-52E细胞上OAT1和OAT3的蛋白表达,证实了以上观点[21]。OAT3蛋白表达下调部分导致OAT3的底物组胺H2受体拮抗剂法莫西汀在缺血再灌注诱导的急性肾损伤大鼠中肾脏排泄清除率降低,血浆药物浓度升高[20]。在顺铂诱导的急性肾损伤大鼠模型中,血清中OAT3的经典内源性底物硫酸吲哚酚浓度显著升高,伴随着大鼠肾脏OAT1和OAT3的mRNA和蛋白表达水平降低。而给予顺铂造模大鼠硫酸吲哚酚的前体(吲哚)的吸附剂AST-120后,大鼠血浆硫酸吲哚酚浓度显著下降,同时肾脏OAT1和OAT3的mRNA和蛋白表达水平恢复,提示硫酸吲哚酚和OAT1及OAT3的表达有密切关系,具体调控机制有待进一步研究[22]。对顺铂诱导的急性肾损伤大鼠给予OAT1和OAT3底物药物头孢地尼后,头孢地尼药物代谢动力学发生改变,表现为尿液累积排泄量降低,血药浓度升高,这一现象与顺铂诱导的急性肾损伤大鼠肾脏OAT1和OAT3的mRNA和蛋白表达下调一致;OAT1和OAT3底物药物头孢妥仑主要通过胆汁排泄,在此模型中药物代谢动力学不受影响[24]。肾损伤病人在进行抗生素治疗时应选择头孢妥仑,或者减少头孢地尼的用药剂量。另外顺铂会导致肾脏OAT5的蛋白表达呈时间依赖性下调,造模后第2 d在尿液中就检测到大量的OAT5,因此尿液中OAT5的增多可能是急性肾损伤的早期生物标志物,它的出现早于尿素氮、肌酐等肾损伤标志物[23]。准确地建立OAT5的检测手段以及明确OAT5异常变化与肾功能损伤的联系是将OAT5作为肾损伤标志物的一个亟待解决的问题。叶酸诱导的急性肾损伤严重损害肾小球和肾小管,该模型大鼠肾脏OAT1和OAT3的mRNA和蛋白表达下调。OAT1底物贝洛替康在叶酸诱导的急性肾损伤大鼠中肾脏排泄清除率明显降低[25]。甲氨蝶呤在临床上被广泛用于治疗类风湿性关节炎、急性白血病等疾病,有严重的肾毒性。在甲氨蝶呤诱导的急性肾损伤大鼠中,肾脏OAT1和OAT3的mRNA和蛋白表达下调,BCL6浓度下降。体外实验表明,沉默BCL6阻断了NRK-52E细胞上OAT1和OAT3的mRNA和蛋白表达。BCL6在胚胎发育和免疫应答中发挥重要作用,可通过蛋白相互作用抑制NF-κB的活性。这些结果表明甲氨蝶呤诱导的急性肾损伤大鼠通过下调BCL6水平激活NF-κB信号通路,从而下调OAT1和OAT3[26]。
2.1.2 慢性肾损伤 慢性肾功能衰竭是指肾单位慢性进行性不可逆被破坏,残存的肾单位不足以充分排出代谢废物和维持内环境恒定,导致代谢废物和毒物在体内蓄积,病人需要通过定期血液透析来排出毒素。目前已经发现有100多种尿毒素在尿毒症患者体内蓄积,这些尿毒素很多都是OAT1和OAT3的底物[47]。慢性肾损伤会导致OAT的表达和功能发生改变,同时OAT的变化对慢性肾损伤的发展进程也起着关键作用。腺嘌呤诱导的慢性肾损伤模型是一种能够很好地反映电解质异常的模型,该模型大鼠肾脏OAT1、OAT3和URAT1的mRNA和蛋白水平均显著下调,OAT1和OAT3的底物苄青霉素的肾脏摄取清除率明显降低,说明OAT1和OAT3的功能降低[27]。5/6肾切除是常用来模拟人类慢性肾衰竭的大鼠模型。5/6肾切除慢性肾损伤大鼠肾脏OAT1、OAT2、OAT3和URAT1的mRNA和蛋白表达均下调,用该模型大鼠血清培养的人肾皮质近曲小管上皮细胞系(HK-2)得到了与体内研究一致的结果,表明肾损伤大鼠血清中可能存在异常成分调控这些转运体[28]。5/6肾切除也会降低大鼠脑OAT3的mRNA和蛋白表达水平,在用该模型大鼠血清培养的大鼠原代脑微血管内皮细胞和星形胶质细胞中进行的研究结果亦显示OAT3水平下调,与体内研究结果一致[29]。通常在疾病状态下,Wnt/β-catenin信号通路被抑制。体内研究表明,激活大鼠脑β-catenin后,血脑屏障上OAT3蛋白活性升高[48],研究人员猜测慢性肾损伤状态下血脑屏障上OAT3的下调可能与Wnt/β-catenin信号通路被抑制有关。此外,慢性肾损伤导致的多种并发症与OAT转运体有密切关系。如大量文献证明肾损伤状态下会出现行动性震颤、反射亢进、抑郁、烦躁和认知功能障碍等中枢神经系统功能障碍[49]。尿毒素蓄积是引发这一系列中枢神经系统功能障碍的机制之一,大部分尿毒素是有机阴离子,如硫酸吲哚酚、马尿酸、硫酸对甲酚及脑内神经递质代谢产物高香草酸、5-羟基吲哚乙酸、喹啉酸和犬尿喹啉酸等,这些物质均会对神经元产生毒性,可能会直接影响血脑屏障上OAT3蛋白表达和功能,又因这些有机酸都是OAT3的底物[10,50],且血脑屏障上表达OAT3,故这些有机酸类尿毒素可竞争性抑制脑内有机酸小分子的外排,导致脑内有机酸蓄积,产生以上中枢毒性[51]。
2.2 肝损伤
肝脏是外源性物质代谢降解的主要器官,病毒、药物、酒精和肥胖等因素都会引起肝损伤,导致肝脏纤维化,进而肝硬化,最后发展成为肝癌。肝脏上OAT参与胆汁的肝肠循环,其中OAT3在胆汁酸的肠-肝-肾轴的转运中起重要作用,参与了胆汁酸的吸收、代谢和排泄[52]。OAT蛋白的变化最终会引起内源性物质和外源性药物转运速率异常改变,引起血浆中代谢物质的累积。OAT2底物药物恩替卡韦为鸟苷环戊酸类似物,是治疗乙型肝炎的一线药物,其通过抑制乙型肝炎病毒聚合酶活性发挥抗乙型肝炎病毒作用。肝硬化状态下,肝脏OAT2下调,导致恩替卡韦在肝脏浓度降低,影响药物的疗效[53]。有文献报道肝损伤状态下,体内TNF-α、IL-6等炎症因子的异常升高可显著下调肝脏细胞OAT2的mRNA表达水平[33],这可能与药物疗效降低有关。临床研究表明,部分肝癌患者对OAT2底物药物5-氟尿嘧啶耐药,这可能是由于肝细胞OAT2表达下调,肝脏5-氟尿嘧啶浓度降低所致。体外MTT实验结果表明,OAT2的底物5-氟尿嘧啶抑制过表达OAT2的肝癌BEL-7402细胞的IC50是对正常BEL-7402细胞IC50的3倍以上[54]。有研究报道OAT2的表达减少会增加丙型肝炎中肝细胞癌的发生及复发风险,具有显著的临床意义[55]。在肝损伤模型中,造模持续时间和造模方式对OAT1和OAT3的表达调控并不一致。从肝脏流出的胆汁减少或受阻造成胆汁淤积是肝损伤的一种,α-萘基异硫氰酸酯和胆管结扎诱导的是胆汁淤积型肝损伤动物模型,这2种肝损伤诱导方式对肾脏OAT1和OAT3的调控不同——α-萘基异硫氰酸酯会下调肾脏OAT1和OAT3的蛋白表达水平,而胆管结扎3 d的大鼠模型中,肾脏OAT1表达下调,OAT3表达上调[16,30]。相反的是,胆管结扎18 h常被用来模拟急性阻塞性黄疸,该模型大鼠肾脏OAT1和OAT3蛋白表达水平升高,且OAT1和OAT3底物药物呋塞米的排泄显著增多,造成这一现象的原因是急性胆道阻塞大鼠肝功能异常所引起的肾脏OAT1和OAT3表达和功能代偿性增加[31]。另有研究表明,急性胆道阻塞也会上调肾脏OAT5的蛋白表达[32]。
2.3 糖尿病
糖尿病是由多种原因引起的胰岛素分泌不足以及靶细胞对胰岛素敏感性降低,继而引起糖、蛋白质、脂肪及水电解质代谢异常的一种临床综合征。糖尿病作为一种系统性疾病可以引起多种组织、器官转运体蛋白表达和功能的改变。诸多研究表明糖尿病可以改变许多药物的体内药代动力学行为,因此研究糖尿病状态下转运体的表达和功能变化意义深远。研究Ⅱ型糖尿病一般有3种常用的啮齿类动物模型,即ob/ob(肥胖)小鼠、db/db(糖尿病)小鼠,以及喂食高脂饲料并给予低剂量链脲佐菌素的大鼠。在ob/ob小鼠模型中,雌性和雄性小鼠肾脏OAT2的mRNA水平均显著低于正常小鼠[34]。db/db小鼠肾脏OAT1和OAT2的mRNA也显著下调[35]。相反的是,高脂喂养合并链脲佐菌素诱导的大鼠模型肾脏OAT2的蛋白表达上调了2倍。高水平的OAT2会增加药物在肾脏的蓄积,进而增加具有肾脏毒性药物的不良反应[36]。
在单剂量腹腔注射链脲佐菌素诱导的Ⅰ型糖尿病大鼠肾皮质中,硫酸雌酮摄取减少与OAT3的表达降低有关,而胰岛素和阿托伐他汀联合治疗可逆转OAT3表达和功能的下调,并伴随丙二醛(malondialdehyde,MDA)、超氧化物歧化酶(superoxide dismutase,SOD)、核因子E2相关因子2(nuclear factor erythroid 2-related factor 2,Nrf2)水平恢复正常,提示可能是胰岛素治疗消除了高血糖导致的氧化应激[37]。体外实验也表明胰岛素可浓度依赖性上调人cos-7细胞上OAT4的蛋白表达和转运功能[40],提示糖尿病状态下OAT表达和功能下调可能与胰岛素缺乏有密切关系。OAT1和OAT3的底物药物如呋塞米和苄氟噻嗪通过影响肾脏近端小管上皮细胞显示其利尿作用。多项研究表明,OAT1或OAT3缺乏会损害呋塞米和苄氟噻嗪的利钠作用,推测糖尿病引起的OAT表达和功能降低可能会削弱利尿剂的利尿作用。在OAT3-/-小鼠和OAT1-/-小鼠模型中,呋塞米和苄氟噻嗪的剂量-尿钠曲线均发生显著右移,呋塞米在OAT3-/-小鼠中钠排泄的EC50是野生型小鼠的3倍,苄氟噻嗪在OAT3-/-小鼠中钠排泄的EC50是野生型小鼠的2.5倍;呋塞米在OAT1-/-小鼠中钠排泄的EC50是野生型小鼠的5倍,苄氟噻嗪在OAT1-/-小鼠中钠排泄的EC50是野生型小鼠的4倍[38]。与此推论一致,托拉塞米的利尿效率在四氧嘧啶诱导的糖尿病大鼠和链脲佐菌素诱导的糖尿病大鼠中均显著降低[39]。这些结果也可以解释糖尿病患者需要更高剂量呋塞米的临床发现。在Ⅰ型糖尿病Ins2Akita小鼠模型中,小鼠肾脏OAT1、OAT3和OAT5的mRNA表达水平显著降低,OAT3蛋白表达水平显著降低[41]。OAT1/3底物恩格列净是钠-葡萄糖协同转运蛋白2(SGLT2)抑制剂,在OAT1-/-和OAT3-/-小鼠中,恩格列净的降糖作用显著低于其对正常小鼠的作用,这表明肾脏OAT3的下调会降低恩格列净的降糖作用[56]。另有动物实验结果表明,抑制肾素-血管紧张素-醛固酮系统可增加OAT3的表达及有机阴离子的清除率,这与血管紧张素转化酶抑制剂治疗的糖尿病病人有机阴离子马尿酸盐的清除率增加的结果一致[57]。进一步研究表明,血管紧张素Ⅱ下调人cos-7细胞上OAT3膜蛋白表达,抑制OAT3底物硫酸雌酮的摄取,而PKC抑制剂可以逆转这一过程,证明血管紧张素Ⅱ通过激活PKC通路下调OAT3蛋白表达[58]。这一过程是通过泛素化连接酶Nedd4-2磷酸化实现的,Nedd4-2磷酸化可以增加OAT的泛素化,加速OAT从细胞膜转向胞内进而被降解。PKC激活对OAT的调控存在时间差异,短期激活PKC,加速了OAT蛋白的泛素化,进而导致OAT蛋白的内吞加速,表现为总蛋白水平不变,膜蛋白水平降低,转运活性下降;长期激活PKC,可促进蛋白酶体途径对OAT的降解作用,表现为总蛋白水平降低,转运活性降低[59]。
2.4 其他疾病
急性肾损伤是急性心肌梗死的常见并发症,心肌缺血再灌注诱导的心肌梗死大鼠肾功能受损伴随着OAT1和OAT3转运功能下降,OAT1功能下降可能会进一步加重肾损伤,这可能是预防急性心肌梗死后肾功能损伤的一个潜在治疗靶点[43]。阿尔茨海默病患者肾功能受损,肾小球滤过率降低。蛋白质组学研究结果表明阿尔茨海默病小鼠肾脏药物转运体表达和功能发生显著变化,其中肾脏OAT3蛋白表达水平升高1.3倍,这可能会影响作为转运体底物的药物和代谢物的清除[42]。高尿酸血症是一种常见的代谢性疾病,尿酸水平的显著升高与痛风、肝肾功能障碍、全身炎症、心血管疾病的发生有关。氧嗪酸诱导的高尿酸血症小鼠其肾脏OAT1的mRNA和蛋白表达水平显著降低,URAT1的mRNA和蛋白表达水平显著升高,OAT3的蛋白和mRNA水平无变化[44]。高尿酸血症大鼠肾脏OAT1和OAT3的mRNA和蛋白表达水平均降低,伴随着OAT1和OAT3底物甲氨蝶呤和头孢氨苄血药浓度的升高[45]。
3 有机阴离子转运体介导的药物相互作用
由于OAT底物的广泛性,该转运体可能会引起药物相互作用,尤其是以肾OAT1或OAT3介导消除为主的药物。FDA指南建议,候选药物原型的肾主动分泌清除率大于或等于药物总清除率的25%时,要通过体外过表达OAT1/3的CHO细胞株、HEK293细胞株或MDCK细胞株评估该药物是否为OAT1/3的底物和抑制剂。如果候选药物在转染OAT1或OAT3的细胞中的摄取率是在对照细胞(或含有空白载体的细胞)中的2倍及以上,或OAT1或OAT3的已知抑制剂在高于其抑制常数(Ki)或者IC50至少10倍的浓度时,使候选药物的摄取降低至50%以下,这提示候选药物可能是OAT1或OAT3的底物。采用过表达OAT1/3的细胞株考察候选药物对已知OAT1/3底物摄取的抑制能力,如果OAT1或OAT3的候选药物稳态下最大游离血浆浓度(Imax,u)与IC50比值≥0.1,则候选药物可能会在体内抑制这些转运体的功能。若体外实验表明候选药物是OAT1或OAT3的底物和抑制剂,应基于安全性和有效性考虑是否进行临床药物相互作用研究[60]。有研究报道,β内酰胺酶抑制剂阿维巴坦的肾脏清除率占总清除率的84.6%;利用过表达OAT1或OAT3的HEK293细胞株研究阿维巴坦的摄取,结果表明阿维巴坦是OAT1/3的底物[61]。另有文献报道,质子泵抑制剂兰索拉唑与抗叶酸药物培美曲塞均通过肾脏OAT3分泌消除,兰索拉唑通过抑制培美曲塞的肾脏消除,增加培美曲塞的血浆浓度,进而加重培美曲塞的血液毒性[62]。又如OAT3底物阿昔洛韦在尿液中溶解度低,容易在肾小管形成结晶,OAT3抑制剂丙磺舒通过减少阿昔洛韦的肾清除,从而降低阿昔洛韦在肾中的暴露量,减轻阿昔洛韦的肾小管结晶不良反应[63]。OAT对药物的体内处置有重要作用,肝、肾功能不全或患有糖尿病等疾病的患者除了联合用药时可能会发生药物相互作用以外,这些疾病本身引起的肾脏OAT1/3功能和表达的变化,也会显著影响OAT1/3的底物药物如青霉素、头孢地尼的药物代谢动力学行为[24,27],因此疾病状态下基于转运体介导的药物相互作用的研究更复杂且意义重大。
4 结语与展望
OAT在组织和体液之间起着运输小分子内源性代谢物、药物和毒素的作用。肾损伤、肝损伤和糖尿病等疾病状态下,肾脏、肝脏和血脑屏障上OAT的表达和功能会发生异常变化。这些变化导致药物经转运体和代谢酶作用时,其体内处置过程不同于正常机体功能下的体内处置过程,导致药物在体内的作用时间和作用强度发生改变,从而产生不良反应甚至严重毒副作用,这增加了药物临床治疗的不确定性,也给临床用药增加了难度。因此需要进一步研究OAT和各种疾病之间的联系,以提高治疗效果,减少可能的毒性。另外,目前有关OAT的研究多停留在动物层面,考虑到种属差异,采用通过基因编辑表达人OAT的动物模型研究疾病状态下OAT功能和表达的改变,以完成从动物数据到人体数据的转化,或将成为未来的研究重点。
