3C样蛋白酶抑制剂博福特韦的研发实践和策略
2023-08-22谢东胡敏姚成
谢东,胡敏,姚成
(前沿生物药业(南京)股份有限公司,江苏 南京210012)
1 博福特韦简介
博福特韦(bofutrelvir,FB2001)为一款3C样蛋白酶(3C-like protease,3CLpro)抑制剂[1],由前沿生物药业(南京)股份有限公司(以下简称前沿生物药业)联合中国科学院上海药物研究所、中国科学院武汉病毒研究所共同开发,用于治疗新型冠状病毒(SARS-CoV-2,以下简称新冠病毒)感染(COVID-19)。团队开发了注射和雾化吸入给药2个剂型,在中国和美国完成了3项Ⅰ期临床试验,目前正在开展Ⅱ/Ⅲ期临床研究。
在药物开发过程中,前沿生物药业研究团队基于不同人群的需求和病毒作用靶点的特点,选择了静脉注射和雾化吸入2个剂型,体现了与口服制剂差异化的开发策略以满足不同的临床需求。新冠病毒变异主要发生在S蛋白,结合3CLpro相对保守的特点以及FB2001的分子结构,研究团队进行了一系列体外和体内抗病毒研究,充分评价了FB2001对多种冠状病毒及不同亚型的活性,并开展了体外耐药病毒培养和敏感性鉴定,结果显示FB2001具有广谱抗病毒活性和高耐药屏障,同时证实了FB2001在不同给药途径下对感染SARS-CoV-2的转基因小鼠的体内抗病毒活性。基于模型引导的药物研发(model-informed drug development,MIDD)策略,研究团队应用群体药代动力学(population pharmacokinetics)和基于生理的药代动力学(physiologically based pharmacokinetics,PBPK)模型,建立了人体静脉给药PBPK模型并根据血药浓度进行验证和优化,预测人体肺组织FB2001浓度,指导临床剂量选择。面对突发的新型病毒传染性疾病,如何基于既往研发经验、现有指导原则和同类品种的成功经验设计并开展临床试验至关重要,研究团队在受试人群选择、主要终点指标确定、试验设计等方面也进行了一些深入的考量。
2 产品亮点及创新点
2.1 剂型的选择
目前全球已获批上市或获得紧急使用授权(EUA)的抗新冠病毒小分子药物,除了治疗住院的中、重症感染者的瑞德西韦(remdesivir)以外,全部是口服药。口服药具有给药与储备便利的优势,但也存在药物颗粒大、不易吞咽,部分药物生物利用度低、需要联用肝药酶抑制剂利托那韦而对基础病治疗产生药物配伍禁忌,以及新冠病毒感染主要靶器官呼吸道和肺部的药物浓度难以迅速达到有效病毒抑制浓度等方面的挑战。
针对这些问题,为了使COVID-19患者,特别是老人、儿童、服药困难者以及有基础疾病的患者有更多的治疗选择,前沿生物药业开发了FB2001注射和雾化吸入2种给药剂型,预期有以下潜在优势。
1)多项研究显示,住院患者往往不仅在肺部,还在体内多个器官存在病毒复制[2]。FB2001注射制剂单药给药,不需要吞咽,不经胃肠道吸收,可迅速到达全身主要器官。FB2001对CYP450无明显抑制作用,其对CYP2C8、CYP2C19和CYP2D6的IC50均大于100 μmol · L-1,对CYP1A2、CYP2B6、CYP2C9和CYP3A4/5的IC50大于20 μmol · L-1,此外, FB200130 μmol · L-1对CYP1A2、CYP2B6和CYP3A4酶活性没有诱导作用,因此,临床上FB2001不需要联用肝药酶抑制剂,也就不会影响到合并症治疗药物的使用。研究团队目前正在开展FB2001注射制剂治疗新冠感染中、重症住院患者的临床研究。
2)COVID-19的主要靶器官为呼吸道和肺部,雾化吸入制剂可以将药物直接递送至这些组织,局部药物暴露量(AUC)高,药物起效快,不需要吞咽,无需联用肝药酶抑制剂,因此不影响合并症治疗药物的使用;与此同时,血液和全身系统AUC低,药物安全窗大。研究团队目前正在开展FB2001雾化吸入制剂治疗COVID-19轻、中症患者的临床研究。
2.2 药物肺部分布显著高于血液,有效清除肺部病毒
研究显示,FB2001经静脉注射和雾化吸入后在鼻腔、气管和肺部的AUC显著高于血液。静脉注射24 h后,FB2001在大鼠和犬肺部药物浓度分别约为血浆药物浓度的83倍和131倍,雾化吸入FB2001后24 h,大鼠肺部药物浓度超过血浆药物浓度的450倍,肺组织中AUC也达到血浆的66倍。
在分别感染了德尔塔株和奥密克戎株的K18-hACE2转基因小鼠中进行的研究结果显示,FB2001经腹腔注射、雾化吸入和滴鼻给药可明显降低小鼠脑部和肺部的病毒载量和病毒滴度,给药后第4 d肺部和脑部病毒滴度可下降至检测限之下[3]。
2.3 广谱抗冠状病毒活性和较高耐药屏障
FB2001具有广谱的抗冠状病毒活性,除了对新冠病毒的3CLpro具有抑制作用外,对SARSCoV-1、MERS-CoV、HCoV-NL63、HCoV-OC43、HCoV-229E和HCoV-HKU1等多种冠状病毒的3CLpro也显示抑制作用。
随着奈玛特韦(nimatrelvir)和瑞德西韦在全球的批准和广泛使用,病毒耐药的发生不可避免,关于其耐药病例不断被报道[4-5]。因此,研究团队在早期就开展了一系列关于FB2001的耐药研究,结果表明新冠病毒在FB2001药物压力下体外培养20代未筛选出耐药突变株。一项研究显示,FB2001对奈玛特韦和恩赛特韦(ensitrelvir)耐药新冠病毒依然保持着较高的活性,具有更高的耐药屏障。M49L、E166A、E166V和L167F单突变株,以及L50F/E166A、L50F/E166V和ΔP168/A173V多突变株对奈玛特韦和/或恩赛特韦具有高度或中度耐药,而对FB2001仍保持敏感或低度耐药[5]。
3 博福特韦研发经验
3.1 关注毒株变异对药物活性的影响
新冠病毒属于RNA病毒,变异性较高,开发药物需要特别关注变异病毒的产生和演化,以及病毒变异对于药物活性的影响。目前抗新冠病毒药物主要为靶向S蛋白的抗体药物,以及靶向RNA依赖性RNA聚合酶(RdRp)和3CLpro的小分子药物。
在病毒自然演化以及疫苗压力下,新冠病毒已经历了从原始毒株到阿尔法毒株、贝塔毒株、德尔塔毒株、奥密克戎毒株的变异,并多次造成大流行,这些毒株的主要突变呈现在S蛋白,使得大部分抗体药物失效,目前几乎所有获批临床的抗体药物对现流行的奥密克戎毒株失去了有效的中和活性[6]。这些流行毒株含有的突变,有些发生在RdRp和3CLpro基因序列,但目前对小分子药物影响较小。随着小分子药物大规模使用,我们需要高度关注在药物压力下耐药病毒的产生和选择。例如,已有对瑞德西韦临床耐药毒株的报道[7]。另外,实验室研究显示,在GISAID数据库收集的流行新冠病毒序列中,有多个对奈玛特韦和恩赛特韦耐药[4]。
3.2 模型引导的药物研发在博福特韦开发中的应用
MIDD通过采用建模与模拟技术对生理学、药理学以及疾病过程等信息进行整合和定量研究,从而指导新药研发和决策。近年来,国内外监管机构发布了多个MIDD的技术指导[8],鼓励药品研发企业从事模型分析工作的专业人员在药物研发过程中尽早介入,参与研究设计和数据分析,形成MIDD模式,提高研发效率。
在FB2001的研发过程中,前沿生物药业研究团队较早地引入了MIDD辅助临床开发。例如在中美Ⅰ期临床研究中,研究团队开展了群体药代动力学研究,考察不同因素对药代动力学的影响,模拟多种给药方案,为Ⅱ/Ⅲ期研究提供依据。
抗新冠药物研发中,肺部和呼吸道药物的浓度对于临床有效性至关重要。研究显示,新冠病毒主要靶器官在呼吸道和肺部,血液中新冠病毒检出率很低。斯坦福健康中心临床病毒学实验室一项研究表明,6.3%(453/7240)的住院及门诊就诊患者其鼻咽拭子新冠病毒RNA呈阳性,其中85例鼻咽拭子新冠病毒阳性患者的血浆样本新冠病毒检出率仅为32.9%(28/85)[9]。另有研究进行了8136 份临床标本的META分析,结果显示肺泡灌洗液(BLF)标本的新冠病毒检出率最高为 91.8%,血液样本中检出率仅1.0%[10]。鉴于目前技术无法直接测定人体肺组织药物浓度,研究团队根据FB2001化学结构和理化性质,建立了SD大鼠和比格犬静脉给药的PBPK模型,并根据大鼠和犬血药浓度和肺部组织浓度验证并优化模型,采用“学习与确认”循环(learn and confirm cycle),进而建立人体静脉给药PBPK模型并根据血药浓度进行验证和优化,预测人体肺组织FB2001浓度,结合FB2001体外有效浓度估算人体有效剂量(见图1)。结果显示,在临床拟用剂量下,预测肺组织FB2001浓度达到2.5 mg · L-1,远高于其体外EC50(0.019 mg · L-1)[3]。
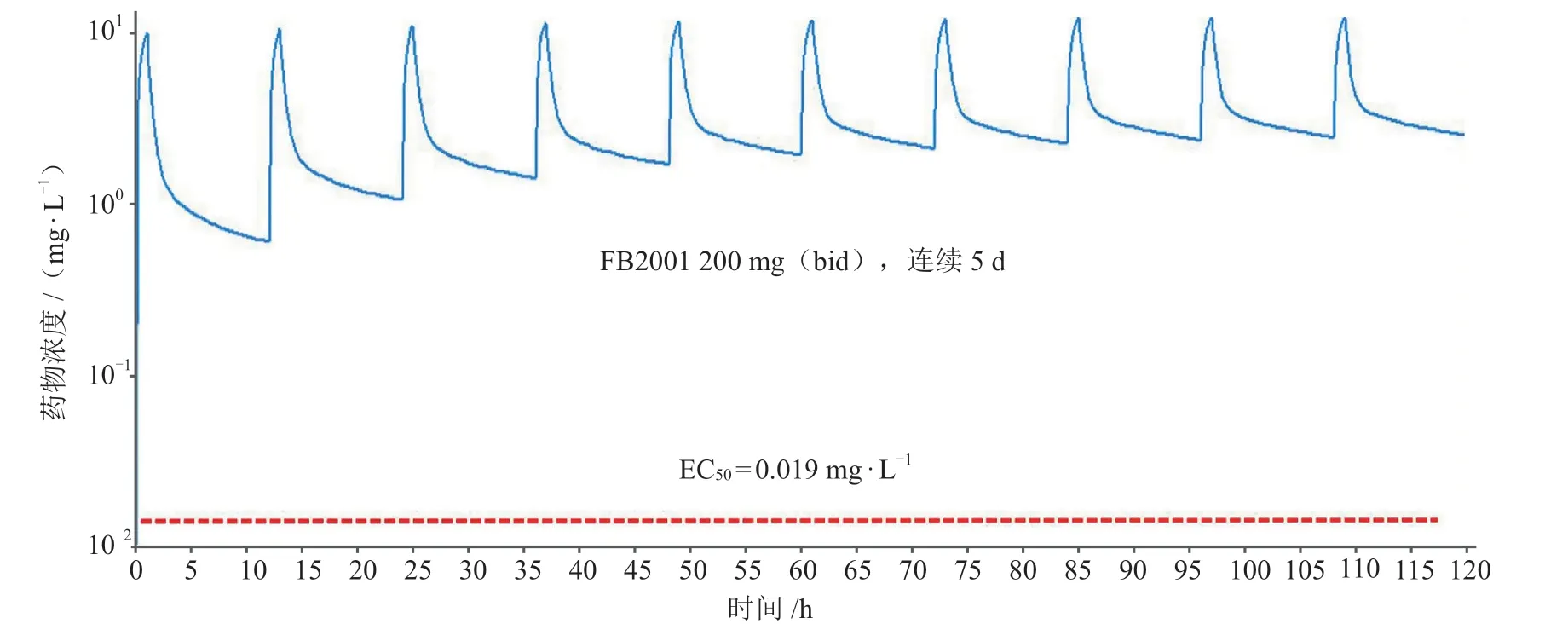
图1 PBPK模型预测的FB2001在肺部的药物浓度Figure 1 Drug concentration of FB2001 in the lung predicted by the PBPK model
4 博福特韦临床研究设计的考量
针对COVID-19,国内外监管机构发布了多个技术指南指导相关药物的临床研究[11-12]。鉴于病毒感染后的动力学特征,一般抗病毒治疗通常考虑尽早给药。在临床研究的设计中,针对不同的新冠感染临床分型(轻、中、重型、危重型),需要制定相应的临床终点。随着新冠病毒在全球的持续传播,新的奥密克戎亚分支将会持续出现,临床研究需要充分考虑新变异病毒株对疾病的影响。
目前还不清楚何种暴露参数或药效学应答参数能够更好地预测药物对COVID-19的疗效,在探索性临床试验中可以将病毒学指标作为主要终点。国内外很多抗COVID-19药物的临床研究将RT-PCR测定的呼吸道病毒载量作为探索性研究的主要终点。而莫诺拉韦(molnupiravir)的Ⅱa期研究中进一步采用了传染性病毒(infectious virus)载量作为终点指标[13],这一指标较一般的病毒载量终点更清晰地显示了药物对活病毒的抑制作用。
对于住院的中重型和/或危重型COVID-19患者的确证性临床研究,主要终点首选适当时间点(如4或8周)的全因死亡率。但是随着病毒变异,全因死亡率评估在实际操作层面存在一定困难,将至临床改善/恢复时间(time to clinical improvement/recovery)作为终点来评估该类药物的临床获益也得到了监管部门的认可。例如瑞德西韦ACTT-1研究[14]、巴瑞替尼ACTT-2研究均将临床状态顺序等级量表定义的至临床恢复时间作为终点指标。
而对于门诊治疗的轻中症患者的确证性临床研究,主要终点首先推荐在适当时间点(至少4周)发展为重型/危重型的发生率或全因死亡率作为主要疗效终点。例如奈玛特韦的EPIC-HR(高风险因素)研究采用门诊患者的住院和全因死亡率作为主要终点指标并获得批准[15]。然而同样随着病毒变异,重症发生率和全因死亡率评估在实际操作层面存在一定困难,在适当的时间内评估至持续临床恢复的时间也被认为是可以接受的。值得注意的是,入组门诊轻中症患者的奈玛特韦的EPIC-SR(标准风险因素)研究,主要终点采用至持续症状恢复时间,并未获得成功。而采用该终点的恩赛特韦的Ⅲ期临床,试验组与对照组的恢复时间差异显示出了统计学意义。
上述研究一般采用随机、双盲、安慰剂平行对照,以公认的背景治疗为基础的优效性比较加载设计。但是这些临床研究在不同地区和时期开展,流行病毒株、患者背景和发病窗口不同,所以获得的结果也不尽相同。在FB2001临床研究设计中,我们参考了国内外同行的研发经验,在临床研究方案中考虑了下列重要因素的影响:流行毒株的致病特征、患者发病窗口期、呼吸道病毒载量水平、疫苗接种状况、是否伴有高风险因素(例如老年人,伴有心血管疾病、呼吸系统疾病、糖尿病、慢性肾脏病等基础疾病的人群,免疫功能低下者等)。此外,由于各国防疫政策动态调整,其医疗实践带来相应变化,这也是临床研究的设计和实施面临的重大影响因素。
5 本领域未来发展方向
新冠病毒以及其他可感染人类的冠状病毒在与人类长期共存的过程中,不排除有高致病性、新的冠状病毒出现的可能,因此开发高效、广谱抗新冠病毒药物进行治疗、预防以及战略储备,具有重要意义。
另外,新冠病毒在感染人类与不同动物宿主(尤其免疫力低下群体)并长期活跃的过程中,由于RNA病毒容易发生突变,可能会使得我们现有的疫苗和药物失去保护作用。对此,我们需要在全球范围建立病毒突变监测体系。
对于病毒复制和致病机制、现有药物对特殊人群的治疗疗法、现有药物的组合使用等,尚有很多有待深入研究之处。
6 结语
新冠疫情具有传染快、规模大、变异多的特点,全球各国防疫管控政策、病毒变异情况,以及疫苗种类和接种情况不同,给COVID-19治疗药物的开发带来了巨大挑战。已获批上市或获得紧急使用授权的抗病毒药物给防疫工作提供了有力武器,但由于其关键性临床试验受当时疫苗接种、背景治疗以及当时流行病毒株的局限,在未来新冠疫情中是否仍然能够发挥有效的治疗作用,有待更多高质量循证医学数据的支持。这些研究,也将为人类应对未来可能出现的新型病毒和疫情提供宝贵的经验和技术基础。
致谢:
FB2001项目得到国家重点研发计划公共安全风险防控与应急技术装备(No.2021YFC0864900)、江苏省工业和信息产业转型升级专项资金、南京市科技计划项目-新型冠状病毒传染应急防治(No.ZX20200007)资助。感谢中国科学院上海药物研究所蒋华良院士、柳红教授,中国科学院武汉病毒研究所张磊砢教授,FB2001临床试验的主要研究者张文宏教授、金荣华教授、卢洪洲教授和全体研究人员,感谢前沿生物药业金洁为本文查阅资料和校对文稿。
