新型冠状病毒感染治疗药物研究进展
2023-08-22朱峻良戴权朱韧龙思会余文颖
朱峻良,戴权,朱韧,龙思会*,余文颖
(1. 中国药科大学江苏省生物活性天然产物研究重点实验室,江苏 南京210009;2. 武汉工程大学化工与制药学院绿色化工过程教育部重点实验室,湖北 武汉 430205)
1 引言
自2007年颁布了管理全球卫生应急措施的《国际卫生条例》以来,世界卫生组织(WHO)共宣布了6次“国际公共卫生紧急事件”,其均由RNA病毒引起。冠状病毒也属于RNA病毒,具有较高的变异能力,目前有7种能感染人的冠状病毒,分别为人冠状病毒229E(hCoV-229E)、人冠状病毒NL63(hCoV-NL63)、人冠状病毒OC43(hCoV-OC43)、人冠状病毒HKUI1(hCoV-HKUI1)、严重急性呼吸综合征冠状病毒(SARS-CoV)、中东呼吸综合征病毒(MERS-CoV)和严重急性呼吸综合征冠状病毒2(也称新型冠状病毒、SARS-CoV-2)。其中SARS-CoV、MERS-CoV和SARS-CoV-2对人类危害较大,且造成的并发症严重威胁人类的生命安全,此次新型冠状病毒感染(COVID-19)疫情便是由SARS-CoV-2引起[1]。
自COVID-19疫情发生以来,世界各国人民的生命安全遭受到严重威胁,SARS-CoV-2经过多次突变,现流行的突变株已逐渐成为奥密克戎变种的亚型XXB.1.16和XBB.1.5,与之前的突变株相比,其具备传播速度更快和免疫逃逸能力更强的特点。
2023年5月8日,国务院联防联控机制召开新闻发布会,介绍了COVID-19疫情不再构成“国际关注的突发公共卫生事件”后的防控工作。虽然COVID-19疫情不再构成“国际关注的突发公共卫生事件”,但并不意味着COVID-19疫情作为全球健康威胁已经结束,病毒正在不断变异,疾病危害仍然存在;我们需要高度关注高龄老人,以及没有接种疫苗的、有基础病的人群,因为这类人群还没有脱离首次或二次感染后进展为重症的风险。
回首过去,人类与RNA病毒进行了几十年的抗争;立足今日,SARS-CoV-2对我们的威胁逐渐消散,但似乎是病毒自身的不断变异,促使人类拥有了相对的免疫力,从而赢得暂时的胜利;展望未来,面对下一次与RNA病毒的抗争,我们能否根据以往经验,设计出能够治疗病毒感染的有效药物?因此,对COVID-19治疗药物的研发不应停止,反而需要更进一步的探索,从而启示未来的抗病毒药物研发,让人类能够真正地战胜RNA病毒。
本文介绍了SARS-CoV-2感染机制,并综述了在研的COVID-19治疗药物研究进展,旨在为相关新药的进一步研发提供参考。
2 SARS-CoV-2感染机制
SARS-CoV-2感染主要是由病毒刺突蛋白(spike protein,S蛋白)的S1亚基与血管紧张素转化酶2(ACE2)结合引起的内吞过程(见图1)[2]。SARS-CoV-2的S蛋白拥有一个独特的S1/S2弗林蛋白酶(furin)可识别位点,在S1亚基与ACE2结合后的融合过程中起着关键作用[3]。S1亚基与ACE2结合后,S蛋白融合体需要用蛋白酶如呋喃酶、胰蛋白酶和跨膜丝氨酸蛋白酶2(TMPRSS2)进行裂解,以促进膜融合过程[4]。在病毒进入细胞并解除包膜后,SARS-CoV-2在细胞内溶解,将核衣壳蛋白(N蛋白)和病毒基因组RNA释放到细胞质中,用于翻译病毒基因组的开放阅读框(ORF1a和ORF1b)和合成2种大型复制酶多聚蛋白(pp1a和pp1ab)[5]。与ORF1b的翻译规则相反,pp1a与pp1ab的C端扩展形式是通过核糖体移码产生的,翻译的核糖体从ORF1a阅读框转移到ORF1b阅读框[6]。一般认为这种核糖体移码是为了调节pp1a和pp1ab的比例[7]。然后,ORF1a编码的蛋白酶自动分解pp1a和pp1ab蛋白,产生16种非结构型蛋白(nsp)。木瓜蛋白酶样蛋白酶(PLpro)识别并特异性切割pp1a或pp1ab的裂解位点Nsp1/2、Nsp2/3和Nsp3/4,产生nsp1 ~4[8],其余的裂解位点由3C样蛋白酶(3CLpro或Mpro)识别并切割,产生nsp5 ~ 16[9]。RNA依赖性RNA聚合酶(RdRp,也称nsp12)和解旋酶(nsp13)一起形成复制-转录复合物(RTC)。RTC的形成有利于冠状病毒的复制和亚基因组RNA的合成,这套亚基因组RNA编码了几种结构和附属蛋白[10]。RTC的疏水结构域附着在内质网的限制膜上,从而产生冠状病毒复制结构[11]。然后,全长的正链基因组RNA被转录成全长的负链模板以合成新的基因组RNA,同时合成重叠的亚基因组负链模板。亚基因组mRNA随后被合成,并翻译产生4个结构蛋白和6个附属蛋白[12]。翻译之后,病毒组装迅速进行,膜蛋白(M蛋白)、包膜蛋白(E蛋白)和S蛋白插入内质网。这些蛋白沿着分泌途径迁移到内质网-高尔基体中间室后,与核衣壳和基因组RNA组装形成的螺旋状核衣壳多聚体相互作用而形成成熟的病毒颗粒。最后,含有病毒的囊泡与质膜融合,通过细胞外渗释放[13]。
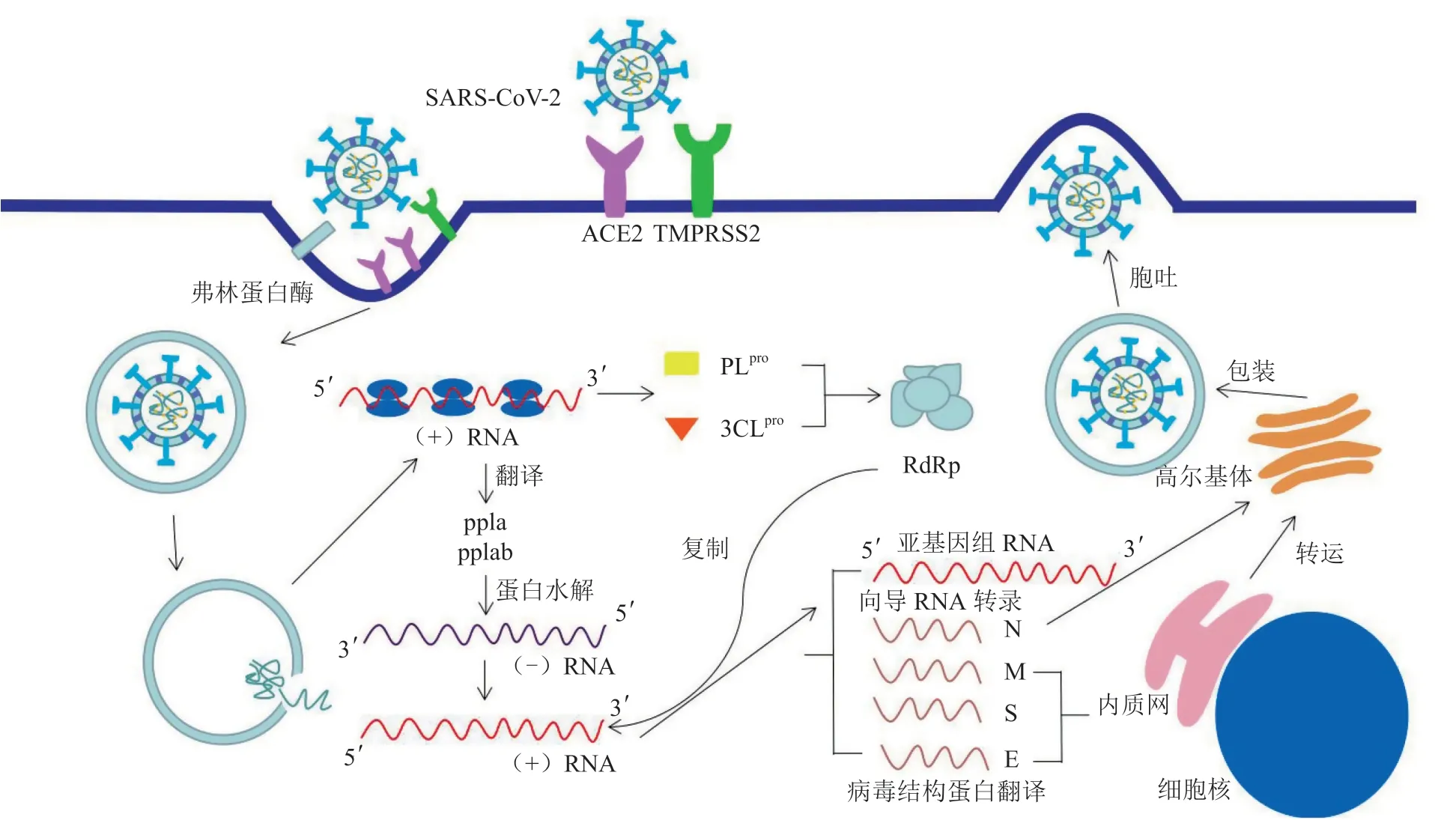
图1 SARS-CoV-2感染宿主细胞的机制Figure 1 Mechanisms by which SARS-CoV-2 infects host cells
S蛋白的中和抗体可阻断S蛋白受体结合域(RBD)与ACE2结合,阻止病毒感染[14]。TMPRSS2已被证明参与病毒入侵过程,靶向该蛋白的药物可能是抑制病毒进入的有效方案[15]。Mpro和PLpro是pp1a和pp1ab转化为nsp的关键蛋白酶,因此对病毒复制十分重要;另外,PLpro的结构类似于人体的去泛素化蛋白酶,其介导抗病毒蛋白去泛素化和去泛素样蛋白15(ISG15)化,从而诱导人巨噬细胞分泌ISG15,而细胞外非偶联的ISG15可发挥细胞因子样作用,反馈性地刺激巨噬细胞,加剧新冠病毒引发巨噬细胞释放大量炎症细胞因子、诱导炎症损害,故PLpro亦是新冠病毒影响宿主固有免疫的关键。因此,抑制该靶点有两方面作用,一是抑制病毒复制,二是抑制病毒引起的炎症,解除病毒对宿主的免疫干扰作用。
3 可作用于病毒靶标的药物
3.1 RNA依赖性RNA聚合酶抑制剂
RdRp与RNA合成密切相关,由于其明确的作用机制和高度保守的特点,能够为药物开发带来两大潜在益处:一方面,能够开发出有效的RdRp抑制剂,不惧新冠病毒突变;另一方面,参照过往抗病毒药物研发历程,不少具有RdRp靶向作用的老药或许能够用于COVID-19治疗,极大地缩短研发周期。因此,RdRp一直是抗RNA病毒药物研发的热门靶点[16]。
疫情初期,为了第一时间发现能用于治疗COVID-19的药物,老药新用成为首选的策略。由于药物的开发过程漫长而代价高昂,重新利用已知药物治疗COVID-19是公认的一种快速而有效的方法[17]。许多用于抗SARS-CoV和MERS-CoV的已上市或在研药物及其他FDA批准的抗病毒药物都被用于抗SARS-CoV-2的研究,尽管取得了一定的进展,但仍需要足够的临床数据来确定其是否真正具有治疗COVID-19的功效[18]。
瑞德西韦(remdesivir)是抗SARS-CoV-2药物研发中老药新用的一个案例,该药由吉利德科学公司开发,用于治疗埃博拉病毒病和马尔堡病毒感染,后来研究人员发现其对SARS-CoV-2也有很好的疗效。瑞德西韦于2020年10月22日成为第一个获得FDA批准的用于紧急治疗COVID-19的药物,通过干扰RdRp来发挥抗病毒作用[19],其给药方式为静脉注射。瑞德西韦在SARS-CoV-2感染的Vero E6细胞系中的半数最大有效浓度(EC50)为6.6 μmol ·L-1,半数细胞毒性浓度(CC50)大于100 µmol · L-1,选择性指数(SI)为15。在SARS-CoV-2感染的Calu-3细胞中,EC50为0.42 μmol · L-1,EC90为1.08 μmol · L-1[20]。然而多项临床研究表明,瑞德西韦对总体死亡率、机械通气时间和住院时间没有影响[21]。
由默沙东公司开发的molnupiravir(EIDD-2801/MK4482),于2021年11月4日获英国药监局批准上市,成为全球第一款获批用于治疗成人轻度至中度COVID-19的口服药物。该药物是一种核糖核苷类似物,通过抑制RdRp而显示出强大的抗SARSCoV-2活性[22]。Ⅰ期临床试验表明,molnupiravir作为一种新型口服抗病毒药物,在健康受试者中有很好的耐受性和安全性[23]。在Ⅱ期临床试验中,轻度至中度COVID-19患者接受为期5 d的molnupiravir治疗,结果表明该药可降低SARS-CoV-2的转录并成功阻止SARS-CoV-2的复制和COVID-19的进展[24]。Ⅲ期临床试验结果显示,molnupiravir治疗组患者的重症/病死率降低了50%[25]。该药已于2022年12月30日在我国获批。
阿兹夫定(azvudine,FNC)片是我国自主研发的口服小分子COVID-19治疗药物。该药由郑州大学常俊标教授等人研发,通过抑制RdRp而抑制SARSCoV-2在体内的复制。一项临床试验表明,阿兹夫定有助于患者更快康复,缩短核酸转阴时间[26]。另一项临床研究表明,阿兹夫定几乎治愈了所有的COVID-19患者,并显示出比瑞德西韦更好的治疗效果[27]。2022年7月25日,国家药品监督管理局应急附条件批准阿兹夫定片新增适应证,用于治疗普通型COVID-19成年患者。
由我国创新药企君实生物研发的口服抗病毒药物VV116,通过其核苷三磷酸形式靶向病毒RdRp而发挥作用,于2021年11月在乌兹别克斯坦获批用于治疗COVID-19患者。该药在SARS-CoV-2感染的Vero E6细胞系中的IC50为0.67±0.24 μmol · L-1,EC50为0.39 μmol · L-1,并且可以明显减少感染小鼠体内的病毒滴度,改善炎症[28]。Ⅰ期临床试验表明,VV116在健康受试者中表现出良好的安全性和耐受性[29]。该药已于2023年1月29日在我国获批用于治疗轻中度COVID-19患者。
Schäfer等[30]研发了一种RdRp抑制剂GS-621763,其被证实在SARS-CoV-2感染的细胞系和人类原代细胞培养物中具有抗SARS-CoV-2活性,且呈剂量依赖性抑制病毒复制。
3.2 3C样蛋白酶抑制剂与木瓜蛋白酶样蛋白酶抑制剂
在所有冠状病毒中,PLpro和3CLpro对于从pp1a和pp1ab的氨基末端释放几种nsp非常关键,故这2种蛋白酶对SARS-CoV-2的复制和翻译都至关重要。
3.2.1 3C样蛋白酶抑制剂 由辉瑞研发的复方制剂Paxlovid,每粒由300 mg nirmatrelvir(PF-07321332)和100 mg利托那韦(ritonavir)组成。Nirmatrelvir是一种3CLpro的强效抑制剂,能有效抑制SARSCoV-2与相关变异株以及其他冠状病毒的体外复制,可治愈感染了SARS-CoV-2 贝塔或德尔塔变异株的金仓鼠[31]。Paxlovid于2021年12月获得美国FDA的紧急使用授权,截至目前,已在全球10个国家被批准或被授权紧急使用。与安慰剂相比,症状出现后3和5 d内启动Paxlovid治疗,可分别将住院或死亡风险降低89%和88%[32]。
由日本盐野义制药(Shionogi)开发的S-217622是一种口服非共价、非肽的3CLpro抑制剂的临床候选药物。该药在感染SARS-CoV-2的小鼠中显示了出色的口服生物利用度和显著的疗效[32]。Ⅱ期临床试验结果显示,连续服用S-2176223 d,80%的受试者其SARS-CoV-2检测呈阴性;连续服药5 d后,100%的受试者其体内SARS-CoV-2完全消失[33-34]。
除了以上已经完成临床试验或正在进行临床研究的药物,还有多个候选化合物取得了不同程度的进展。例如:Rathnayake等[35]研发的GC376及其衍生物为SARS-CoV-2的3CLpro抑制剂,在Vero E6细胞系和小鼠模型中均可发挥抗病毒作用,是一种潜在的抗SARS-CoV-2药物;Jin等[36]研发的3CLpro抑制剂卡莫氟(carmofur),其脲基可与3CLpro活性位点中的Cys145共价结合,共价结合后其脂肪侧链将占据疏水性S2亚位点;Hasnain团队研发了依布硒(ebselen)及其衍生物,并探索了该类化合物与3CLpro的结合模式,结果表明这些化合物对SARS-CoV-2具有潜在抑制作用[37]。
3.2.2 木瓜蛋白酶样蛋白酶抑制剂 PLpro是pp1a和pp1ab转化为nsp的关键蛋白酶,对病毒复制十分关键;另外,PLpro的结构类似于人体的去泛素化蛋白酶,还可以从信号蛋白中去除宿主泛素和ISG15以抑制先天免疫反应,因此,抑制该靶点不仅可以抑制病毒复制,还可抑制病毒引起的炎症,解除病毒对宿主的免疫干扰作用。
3.3 解旋酶抑制剂
nsp13是由病毒基因组编码的具备5'-3'定向解旋酶、ATP酶和RNA 5'-三磷酸酶活性[39]的多功能蛋白质,其与其他nsp协同组装成复制转录复合物,从而参与病毒RNA的复制与转录[40-43],是病毒基因组复制与转录的关键[44];另外,nsp13还具备高度保守的特点,SARS-CoV和SARS-CoV-2的nsp13之间只有单一的氨基酸差异,故其被认为可能是抗COVID-19的一个很有前景的靶点。针对该靶点报道的抑制剂主要有巴拿宁、5-羟基色酮衍生物[45-46]和SSY A10-001[47],其体外细胞活性均在微摩尔级范围,然而由于这些抑制剂可与ATP竞争结合位点,故也能与体内其他具备ATP酶活性的酶结构域进行结合而产生副作用,如何得到选择性良好和活性更佳的抑制剂是该靶点相关药物研发所面临的一大挑战。
3.4 结构蛋白抑制剂
S蛋白是位于病毒表面,形状类似皇冠的结构蛋白,以三聚体形态存在,在病毒感染的初始过程中发挥极其重要的作用。其能被宿主细胞表面的ACE2受体识别并结合,进而完成病毒附着过程。然而,由于编码该蛋白的基因组很容易受到环境的压力而发生变异,极大地升高了药物研发的失败率,故针对该靶点的小分子药物研发并不多,但其却是疫苗和抗体药物研发的热点[17]。
E蛋白是新冠病毒中最小的,也是人们最不了解的一种结构蛋白,它在不同的病毒亚型中高度保守。研究尚未完全阐明E蛋白在病毒入侵、复制和释放中的作用。病毒颗粒包膜中的E蛋白通过与其他结构蛋白相互结合而发挥作用。E蛋白和M蛋白相互作用,维持病毒颗粒的形状并促进其释放[48-49]。缺乏E蛋白会显著降低病毒滴度和子代病毒的成熟度,甚至产生无感染功能的子代病毒[50],这表明E蛋白在病毒产生和成熟中的重要性。针对该靶点的抑制剂有hexamethylene amiloride(HMA)和amantadine(AMT)及两者组合物,HMA和AMT是广泛的病毒通道蛋白抑制剂,除了对冠状病毒有作用外,它们对HIV、鸡传染性支气管炎病毒(IBV)和小鼠肝炎病毒(MHV)也有良好的抑制作用[51-53]。
M蛋白是SARS-CoV-2中含量最多的结构蛋白[54],具有3个跨膜结构域,属于跨膜蛋白[55],其能与E蛋白、S蛋白、N蛋白稳定结合以促进SARS-CoV-2核心结构的组装[56]。
N蛋白主要功能是将病毒基因组RNA包装成长螺旋核糖核酸(RNP)复合物,并通过与M蛋白相互作用而参与病毒粒子的组装[57]。N蛋白参与冠状病毒复制周期和宿主细胞对病毒感染的反应[58],因此,它也是COVID-19药物研发的潜在靶点。相关化合物根据作用机制可以分为以下两类。一类为直接抑制RNA与N蛋白的结合从而阻止RNP的形成。到目前为止,已经有一些针对其他冠状病毒的小分子化合物通过虚拟筛选被认为是SARS-CoV-2的候选抑制剂,例如靶向RNA结合位点、可抑制hCoV-OC43复制的化合物PJ34和H3[59]。另一类为阻断正常N蛋白寡聚化或触发异常RNP形成。我国台湾省中兴大学的候明宏团队通过虚拟筛选得到的5-苄基氧代精氨酸(化合物代号:P3)是一种新型的MERS-CoV抑制剂,该化合物可介导MERS-CoV N蛋白中的N末端域(N-NTD)非天然二聚化并诱导N蛋白聚集。基于结构的研究表明,P3靶向N-NTD间相互结合的界面,并同时与两种N-NTD原聚体中的疏水口袋相互作用[60]。然而这两类小分子抑制剂对SARS-CoV-2是否有效还有待进一步研究。
4 可作用于宿主细胞靶标的药物
4.1 血管紧张素转化酶-2抑制剂
ACE2是一种羧肽酶,全长805个氨基酸,它能从底物的C末端去除一个氨基酸。作为肾素-血管紧张素-醛固酮系统的组成部分,ACE2在正常生理学中的主要作用是将肾素和ACE产生的血管紧张素Ⅰ和血管紧张素Ⅱ分别转化为血管紧张素1-9和血管紧张素1-7。在病毒感染过程中,ACE2通过识别并结合SARS-CoV-2表面的S蛋白而使病毒蛋白附着在细胞表面,进一步通过膜融合进入细胞质,是病毒进入宿主细胞的关键靶点。然而有趣的是,ACE2的催化活性对于病毒的结合并不是必需的,并且催化口袋也不是与病毒的结合位点,因此,ACE2催化位点的传统小分子抑制剂不能阻断SARS-CoV-2的感染[61]。有专家提出模拟人ACE2设计出可溶性重组ACE2蛋白竞争结合S蛋白,以阻断新冠病毒S蛋白与宿主ACE2的结合,从而达到治疗目的。然而,截至目前,还没有此类分子作为抗SARS-CoV-2感染候选药物进入临床试验。
4.2 跨膜丝氨酸蛋白酶2抑制剂
TMPRSS2是一种具有丝氨酸蛋白酶活性的Ⅱ型跨膜蛋白,其主要生理作用和底物特异性尚未明确。尽管如此,它在呼吸道病毒感染中的作用,特别是对SARS-CoV的作用已经得到了很好的证实[62-64]。在SARS-CoV-2感染过程中,病毒S蛋白的S1亚基与ACE2结合后,弗林蛋白酶将S1-S2的连接处切割,露出S2亚基的S2'位点,该位点可被TMPRSS2或体内其他组织蛋白酶识别并切割,从而激活膜融合过程。与内体其他组织蛋白酶的切割途径相比,TMPRSS2切割是SARS-CoV-2感染的首选途径。因此,TMPRSS2抑制剂被认为是治疗COVID-19的潜在药物靶点。
开拓药业(Kintor Pharmaceutical Limited)研发的普克鲁胺(pruxelutamide),可降低ACE2和TMPRSS2的表达而抑制SARS-CoV-2侵入宿主细胞。该药曾在巴拉圭获得紧急使用授权,用于中重度住院COVID-19患者。
近年来,我国应用型本科院校发展取得了一些成绩。在教育部的指导下,以应用技术大学为办学定位的地方本科院校发起并成立了应用技术大学(学院)联盟。截止到2018年6月,已有172所高校加入了该联盟。
小分子化合物N-0385由Jean团队研发,可通过抑制TMPRSS2阻止病毒进入人体。体内与体外研究结果均表明N-0835具有显著的治疗效果[65]。
4.3 弗林蛋白酶抑制剂
弗林蛋白酶是一种1型膜结合蛋白酶,其主要功能为切割新冠病毒S1与S2连接处多碱基位点(Arg-Arg-Ala-Arg),释放出S1亚基,以露出S2亚基的S2'位点,故S1/S2边界的裂解是S2'位点裂解的前提条件[66]。研究人员对弗林蛋白酶切割位点进行定点突变,以探究弗林蛋白酶切割位点是否与SARS-CoV-2感染和致病有关。结果显示,与未突变的病毒相比,突变的病毒复制速度显著下降[67],表明弗林蛋白酶切割位点在SARS-CoV-2感染中的关键作用。弗林蛋白酶抑制剂癸酰-RVKR-CMK已被证明能够有效抑制SARS-CoV-2的感染,然而截至目前,还没有任何这种作用机制的分子进入治疗COVID-19的临床试验。
4.4 组织蛋白酶L抑制剂
尽管大部分情况下SARS-CoV-2被TMPRSS2激活,但S2'位点的切割也可以由组织蛋白酶,特别是组织蛋白酶L介导。如果靶细胞表达的TMPRSS2不足,或者病毒-ACE2复合物没有遇到TMPRSS2,则与ACE2结合的病毒通过网格蛋白介导的内吞作用内化到溶酶体中,S2'位点被组织蛋白酶L切割。设计出具有选择性的组织蛋白酶L抑制剂是抗COVID-19药物研发的另一个思路。Liu团队设计出的MPI8可以选择性抑制SARS-CoV-23CLPro和宿主组织蛋白酶L。因对组织蛋白酶L选择性较高,故MPI8不仅具有良好的抗病毒活性,且对宿主细胞的潜在毒性较低[68]。
5 具有抗SARS-CoV-2活性的化合物研究近况
经文献调研,具有抗SARS-CoV-2活性的部分化合物的体外和体内研究近况见表1,其中大部分化合物抑制活性为微摩尔到纳摩尔级,具有良好的抗病毒活性。

表1 具有抗SARS-CoV-2活性的化合物及其临床前研究情况Table 1 Compounds with anti-SARS-CoV-2 activity and their preclinical studies
6 结语与展望
在过去的几十年里,出现了3种导致人类严重疾病的冠状病毒:SARS-CoV、MERS-CoV和SARS-CoV-2;未来,还可能会有新的病毒出现并威胁人类健康。令人备受鼓舞的是,科学家在SARSCoV-2的传播与发病机制,以及诊断、预防和治疗方面均取得了巨大研究进展,并从以往的抗疫工作中吸取了宝贵的经验教训。因此,在面对未来可能新出现的冠状病毒疫情时,不论是对病毒的生物学探索还是疫苗和治疗方法的开发,都会以前所未有的速度推进。当然,目前该领域仍有许多悬而未决的问题,例如,是什么原因导致了SARS-CoV-2优先进行TMPRSS2切割途径?SARS-CoV-2的变异是否有规律可循?能否用人工智能等手段预测病毒的变异行为,为疫苗和小分子药物的研发提前布局?解决这些问题可能有助于开发能有效对抗冠状病毒的新工具。
笔者所在课题组曾发现一种可同时共价靶向3CLpro和PLpro的活性分子(代号:LY1),已进行了体内和体外抗病毒活性和机制的研究,初期结果显示LY1通过其迈克尔受体位点与3CLpro和PLpro催化活性中心的半胱氨酸残基以不可逆共价的方式结合(见图2),并且对病毒蛋白酶的结合是特异的。LY1对SARS-CoV-2具有显著的抗病毒活性,对3CLpro和PLpro灭活效率良好,急性毒性研究结果显示小鼠和大鼠能较好地耐受该活性分子。后续研究正在进行中。
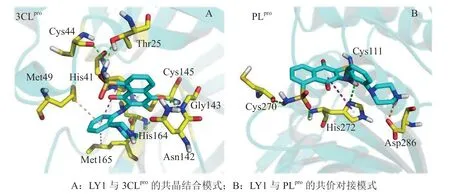
图2 LY1与3CLpro和PLpro的结合模式Figure 2 Binding mode of LY1 with 3CLpro and PLpro
