新型冠状病毒感染治疗药物氢溴酸氘瑞米德韦片的临床研发实践
2023-08-22马娟潘娅丽
马娟,潘娅丽
(上海君实生物医药科技股份有限公司,上海 200126)
新型冠状病毒(SARS-CoV-2,以下简称新冠病毒)感染(COVID-19)是一种由新冠病毒感染宿主引起的疾病,世界卫生组织(WHO)于2020年3月11日宣布其全球大流行。截至2023年6月21日,全球共计报告超过7.6亿例COVID-19确诊病例,死亡人数超过694万。新冠病毒属冠状病毒大家族,是一种单链正股RNA(+ssRNA)病毒,具有单个线性RNA片段[1]。RNA依赖性RNA聚合酶(RdRp)是病毒快速复制的核心蛋白,针对RdRp的抗病毒药物是当前主流研发方向之一[2]。本文介绍了RdRp抑制剂氢溴酸氘瑞米德韦片(商品名:民得维®;代号:VV116或JT001)的临床研发实践,旨在为突发公共卫生事件急需药物的研发提供思路与参考。
1 氢溴酸氘瑞米德韦片特点
氢溴酸氘瑞米德韦片是由上海君实生物医药科技股份有限公司(以下简称君实生物)研发的一种核苷异丁酸酯前药,进入体内后被代谢为核苷形式116-N1,116-N1进一步在细胞内形成具有抗病毒活性的核苷三磷酸活性形式(116-NTP),其通过抑制新冠病毒RdRp发挥抗病毒作用。如图1所示,氢溴酸氘瑞米德韦可靶向新冠病毒RdRp高度保守的活性中心,在病毒复制和转录的环节发挥作用。
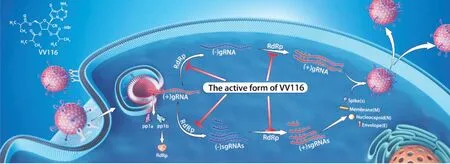
图1 氢溴酸氘瑞米德韦的作用机制Figure 1 Action mechanism of deuremidevir hydrobromide
新冠病毒RdRp的活性中心高度保守,因此以RdRp为靶点的药物有较高的耐药屏障[3]。氢溴酸氘瑞米德韦及其核苷结构116-N1在Vero E6细胞中,对新冠病毒原始病毒株及贝塔、德尔塔、奥密克戎变异株均有抗病毒活性。体外研究结果表明,氢溴酸氘瑞米德韦与经CYP450酶代谢的药物发生相互作用的可能性较低。在人肝微粒体中进行的体外研究表明,116-N1对CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6、CYP3A4/5基本无抑制作用。在人原代肝细胞中进行的体外研究表明,116-N1对CYP1A2、CYP2B6和CYP3A4无诱导作用。用于评价氢溴酸氘瑞米德韦遗传毒性的Ames试验、中国仓鼠肺细胞染色体畸变试验及大鼠骨髓微核试验的结果均为阴性。
关于氢溴酸氘瑞米德韦片的一项已完成的Ⅲ期临床试验(JT001-010)数据[4]及另一项已完成的Ⅲ期临床试验(JT001-015)期中分析数据显示,氢溴酸氘瑞米德韦片(每12 h给药1次,连续5 d,第1 d每次0.6 g,第2—5 d每次0.3 g)相比于安慰剂,可将轻中度COVID-19患者持续临床症状消失时间缩短2 d(氢溴酸氘瑞米德韦片组与安慰剂组分别为10.9和12.9 d),且有显著的统计学差异;伴有进展为重症或死亡的高风险因素的轻中度COVID-19患者,至持续临床恢复时间和至持续症状消失的时间均非劣于奈玛特韦片/利托那韦片组合包装(Paxlovid)。病毒学指标方面,氢溴酸氘瑞米德韦片组新冠病毒的载量、Ct值较基线的变化均优于安慰剂组,至首次病毒转阴时间、转阴比例、Ct值较基线变化与Paxlovid相当。整体不良事件及不良反应发生率与安慰剂组相当,整体不良事件发生率低于Paxlovid。
2 临床研发历程
2023年1月28日,经国家药品监督管理局(NMPA)应急审评审批,氢溴酸氘瑞米德韦片附条件获批上市,距离2021年11月2日获得NMPA药物临床试验批件不到15个月。本项目的快速推进得益于氢溴酸氘瑞米德韦片良好的临床有效性和安全性;此外,及时优化和调整临床试验方案、充分整合研究团队,也发挥了重要的推动作用。
氢溴酸氘瑞米德韦片在我国已完成的临床试验详见表1。

表1 氢溴酸氘瑞米德韦片在中国已完成的临床试验列表Table 1 List of completed clinical trials of deuremidevir hydrobromide tablets in China
2.1 三项Ⅰ期临床试验
为加快研发速度,基于氢溴酸氘瑞米德韦片良好的临床前安全性,君实生物研究团队开展了其单次给药剂量递增(SAD)研究、多次给药剂量递增(MAD)研究,以及食物对药代动力学的影响(FE)研究共3项Ⅰ期临床试验。2021年11月启动氢溴酸氘瑞米德韦片在我国的首次人体试验(first in human,FIH),即SAD研究。同时,君实生物根据临床前研究数据以及SAD研究中已完成的剂量组盲态安全性和药代动力学数据,开始准备MAD研究方案,当SAD研究剂量爬坡到MAD研究中预定剂量并获得其盲态安全性及药代动力学数据后,启动MAD研究。与此同时,FE研究也同步开展。上述3项Ⅰ期临床研究表现出良好的安全性和药代动力学特性,相关数据于2022年3月在学术期刊《中国药理学报》发表[5]。3项研究共入组86例健康受试者,SAD研究显示氢溴酸氘瑞米德韦片半衰期为4.80 ~ 6.95 h;SAD研究中氢溴酸氘瑞米德韦片在25 ~ 800 mg剂量范围内单次给药以及MAD研究中在200 ~ 600 mg范围内多次给药,均显示药物暴露量随剂量成比例增加;FE研究显示标准餐对峰浓度(Cmax)和血药浓度-时间曲线下面积(AUC)无影响。三项Ⅰ期试验均显示良好的安全性:无严重不良事件发生,且未见依据常见不良事件术语评定标准(CTCAE)评为3级及以上的不良事件发生。三项Ⅰ期试验数据支持后续试验的推荐剂量为200 mg 或以上,但不超过 600 mg,每天口服2次,且不超过 5.5 d。
2.2 JT001-010研究
2022年3月底,上海COVID-19疫情防控形势严峻复杂,君实生物研究团队立即开展氢溴酸氘瑞米德韦片对比Paxlovid的非劣效的临床试验(代号JT001-010),该试验为首个与WHO推荐的一线治疗药物Paxlovid进行对比的前瞻性随机对照临床试验。基于当时伴有进展为重症的高风险因素的轻中度COVID-19患者群体特征,研究设计进一步优化,是否接种过新冠疫苗均可入组,主要终点被设定为更适用于奥密克戎变异株疾病特征的“至持续临床恢复的时间”。试验数据于2022年12月在学术期刊《新英格兰医学杂志》(NEJM)[4]发表,试验达到预设终点,氢溴酸氘瑞米德韦片组(每12 h给药1次,连续5 d,第1 d每次0.6 g,第2—5 d每次0.3 g)与Paxlovid组的至持续临床恢复时间中位数分别为4和5 d,风险比(HR)为1.17(95% CI为1.02 ~ 1.36),大于预设的非劣效界值0.8,由于HR的95% CI双侧边界均大于1,试验达到了统计学优效;至持续临床症状消失时间中位数均为7 d(HR为1.06,95% CI为0.91 ~ 1.22);所有受试者均未进展为(危)重症COVID-19或死亡;至首次新冠病毒转阴的时间中位数均为7 d(HR 为0.99,95% CI为0.85 ~ 1.14);氢溴酸氘瑞米德韦片组不良事件发生率低于Paxlovid组(67.4%vs77.3%),整体安全性良好。该试验为单盲设计,研究过程中研究医生和研究团队都是盲态,受试者是非盲态,由于Paxlovid彼时已在中国、美国、欧盟国家等全球多国获批上市且被WHO推荐为治疗轻中度COVID-19的一线药物[6],接受氢溴酸氘瑞米德韦片治疗的非盲受试者有可能更容易主观认为该药效果欠佳或有更多的不良反应,从而对氢溴酸氘瑞米德韦片的疗效或安全性结果产生不利的影响,在这种潜在可能的影响下,试验终点达成更加说明氢溴酸氘瑞米德韦片具有良好的临床有效性和安全性。
2.3 JT001-015研究
不同于JT001-010试验,为满足普通COVID-19患者感染新冠病毒后减轻症状和缩短病程的需求,君实生物启动了JT001-015研究,该试验覆盖更广的患者人群即轻中度COVID-19患者,无论其是否伴有进展为重症或死亡的高风险因素,均有机会入组。试验主要终点“至持续临床症状消失的时间”的设计分别参考了2022年2月NMPA发布的《新型冠状病毒肺炎抗病毒新药临床试验技术指导原则(试行)》[7]中建议的“在轻型和/或普通型新型冠状病毒感染治疗研究中,主要疗效终点也可选择在适当的时间内评估至持续临床恢复的时间”,以及2020年9月美国FDA发布的《COVID-19预防或治疗药物临床试验中门诊成人及青少年受试者COVID-19相关症状评估的指南》[8]。为了保证作为主要终点的受试者自评分数据的真实性和数据质量,研究采用了电子患者报告结局(Epro)设计。君实生物在研究方案的整体设计和重点细节方面与药品审评中心(CDE)的临床和统计专家进行了充分的沟通交流并达成了一致。试验进行过程中经历了2022年12月7日国家防疫政策的重大调整,大多数COVID-19患者不再入住既往的定点救治医院,而是广泛地分布在多数医院的不同科室,这对入组又造成了重大影响。研究团队根据新的COVID-19诊疗路径迅速调整入组策略,不到3个月完成了全部1369例受试者入组。该试验入组迅速归因于君实生物对研究中心的前瞻性布局,为未来的可能入组的中心做好前期准备,同时根据不同中心的情况,指导详细的操作流程,在疫情复杂变化及防疫政策调整的情况下确保了研究质量。期中分析数据显示,氢溴酸氘瑞米德韦片5 d治疗组和安慰剂组根据Kaplan-Meier法估算的至持续临床症状消失(即连续2 d 11项症状评分为0分的第1 d)的中位时间分别为10.9和12.9 d,基于分层的Peto-Peto-Prentice检验结果,氢溴酸氘瑞米德韦片对比安慰剂组生存函数有显著差异(双侧P值为0.0023)。在改良意向性分析集(Mitt)中,所有与症状相关的次要终点,包括至持续临床症状消失(连续3 d 11项症状评分为0的第1 d)的时间和至持续临床症状缓解(连续2 d及连续3 d 11项症状评分不高于1的第1 d)的时间,分析结果均显示氢溴酸氘瑞米德韦片较安慰剂组有所缩短,基于分层的Peto-Peto-Prentice检验结果均有显著性差异。氢溴酸氘瑞米德韦片组的病毒载量及Ct值等病毒学指标较基线的变化均优于安慰剂组,与主要疗效终点趋势一致。安全性方面,氢溴酸氘瑞米德韦片组不良事件和不良反应发生率比安慰剂组更低,未见严重或导致死亡的不良反应。揭盲后,研究团队仅用3 d就完成了新药上市申请材料的撰写和递交。氢溴酸氘瑞米德韦片作为应急药物,审评过程获得了NMPA全力支持,从上市受理到获批仅用了11 d。
3 结语
2023年1月6日,国家卫生健康委员会、国家中医药管理局印发了《新型冠状病毒感染诊疗方案(试行第十版)》[9];3月1日,国家卫生健康委员会将先诺特韦片/利托那韦片组合包装和氢溴酸氘瑞米德韦片纳入《新型冠状病毒感染诊疗方案(试行第十版)》,用于治疗轻、中型COVID-19的成年患者[10]。中国疾病预防控制中心于2023年4月8日发布的《全国新型冠状病毒感染疫情情况》[11]显示,COVID-19患者虽然已大幅减少,但是新冠病毒仍然存在。国产抗新冠病毒药物氢溴酸氘瑞米德韦片附条件获批上市,为国内的COVID-19患者提供了又一治疗方案,也为应对未来的突发情况积累了宝贵的经验与储备。氢溴酸氘瑞米德韦片得益于中科院、科技部、工信部、NMPA等国家各部门的支持以及各个研究中心和研究者们的支持。同时,国内多部委联席的疫情防控机制以及上海市政府各部门联合组建的协调沟通网络使得君实生物的新药研发速度大幅加快。
新冠疫情作为一种突发公共卫生事件,其特殊性决定了COVID-19药物研发不仅仅是做药物研究,还要克服时间紧迫性、毒株不断变异所带来的困难,并根据疫情防控政策的变化及时调整临床研究策略。研发机构要做好前瞻性布局,加强技术和人才储备,形成可迅速响应的应急研发体系和机制,从而能够在更短的时间里完成新药临床研发,以应对新发突发疫情。
