光固化含能粘合剂的设计与合成
2023-08-08袁璟蔺向阳彭洋檀成
袁璟, 蔺向阳, 彭洋,2, 檀成
(1.南京理工大学 化学与化工学院, 江苏 南京 210094; 2.京东方显示技术有限公司, 江苏 南京 210033)
0 引言
复合固体推进剂通常由高分子粘合剂、高能氧化剂、金属燃烧剂和其他助剂均匀混合而成[1-3]。其中粘合剂是最重要的组成成分,虽然其含量通常只占推进剂的10%~20%,但是却决定着推进剂的力学和燃烧等各项性能的表现,是推进剂综合性能能否达标的关键。粘合剂的主要作用是为氧化剂和金属粉末等固体填料提供一种连续均匀的介质,以使这些异相组分之间可以混合均匀,从而保证燃烧的稳定性[4]。另一方面,粘合剂作为一种高分子弹性材料,对各组分还起到包覆作用,赋予了推进剂整体良好的力学性能,可以抵抗一定的外力冲击,保证了在生产、运输、存储和使用等一系列过程中的安全性[5]。因此,寻求综合性能优异的粘合剂一直是研究者们长期奋斗的目标。
聚叠氮缩水甘油醚(GAP)作为一种典型的叠氮类热固性含能粘合剂,具有能量水平高、热稳定性好、生成热高等优点,但也存在低温力学性能不佳、固化成型工艺复杂等问题,近年来对于GAP的改性逐渐成为研究工作的热点[6]。Hafner等[7]将丁基环氧乙烷与GAP共聚,利用非极性的长侧链正丁基来降低聚合物分子链之间的相互作用,从而起到一种分子内塑化的效果,降低了玻璃化转变温度,流体黏度大幅度降低。Mathew等[8]将GAP和端羟基聚丁二烯(HTPB)以不同比例相互混合之后加入二异氰酸酯固化,得到了多种共混弹性体,通过增加催化剂用量加快反应速度的方法改善两组分之间的相容性,并研究了固化剂类型、固化参数R值、GAP含量等因素对共混物力学性能的影响。随着R值的提高,交联密度上升,弹性体的拉伸强度不断提高,而延伸率则在R值为1时下降幅度达到最大,R值继续提高所引起的变化不明显。力学测试表明当GAP含量在30%~50%之间时,具有最高的拉伸强度。Betzler等[9]用GAP作为反应底物,通过对叠氮基团先还原后氧化等一系列合成手段,成功将侧链的叠氮基团转变为硝酸胺基团,得到一种新的含能预聚物聚硝酸胺缩水甘油醚(GNAP),并对其感度和能量水平进行了测试。结果表明,该预聚物的热分解温度和撞击感度均降低至聚硝酸酯缩水甘油醚(PGN)的近似水平,摩擦感度大于360 N,爆速和爆压比PGN要高但略低于GAP。综上所述,目前对于GAP的改性研究主要集中于对其力学性能以及能量特性和感度的优化,而其固化成型工艺复杂的问题并未有效解决。随着光固化3D打印技术的兴起,近年来国内外学者开始尝试用光敏树脂作为一种新型粘合剂,并用其进行推进剂的3D打印研究[10-14]。但使用的光敏树脂大多为以碳氢为结构骨架的惰性有机材料,燃烧所能释放的能量不足,不能满足推进剂对于高能量水平的需求[15-19]。
因此,本文综合考虑GAP含能粘合剂和惰性光固化粘合剂的优势和不足,对于GAP进行端羟基光固化改性,旨在设计并合成一种兼具光固化和高能特性,力学性能接近端羟基聚丁二烯(HTPB弹性体拉伸强度为0.86 MPa,断裂伸长率为240%)的新型光敏树脂作为粘合剂使用,以弥补当前研究领域中的空缺。
本文以环氧氯丙烷为原料,经阳离子开环聚合,叠氮化反应,3种异氰酸酯类化合物、丙烯酸羟乙酯(HEA)亲核加成反应,合成了3种光固化含能预聚物[20-21]。产物结构通过傅里叶红外光谱、核磁共振氢谱和凝胶渗透色谱进行了表征测试。利用高低温差式扫描量热分析对预聚物的热稳定性和玻璃化转变温度进行测试,通过拉伸试验机对其固化膜的力学性能进行了研究。
1 实验部分
1.1 试剂与仪器
试剂包括:二氯甲烷(DCM),规格:分析纯(AR),南京化学试剂股份有限公司产;1,4-丁二醇(BDO),规格:AR,南京化学试剂股份有限公司产;三氟化硼乙醚(BF3Et2O),规格:AR,上海麦克林生化科技有限公司产;环氧氯丙烷(ECH),规格:AR,上海阿拉丁生化科技股份有限公司产;N,N-二甲基甲酰胺(DMF),规格:AR,南京化学试剂股份有限公司产;叠氮化钠,纯度>98%,自制;甲苯(PhMe),规格:AR,国药集团化学试剂有限公司产;二苯甲酮,纯度>99%,上海笛柏生物科技有限公司产;二月桂酸二丁基锡(DBDTL),纯度>95%,上海阿拉丁生化科技股份有限公司产;甲苯二异氰酸酯(TDI),纯度>98%,上海麦克林生化科技有限公司产;对羟基苯甲醚(MEHQ),纯度>99%,上海皓鸿生物医药科技有限公司产;HEA,纯度>99%,上海麦克林生化科技有限公司产;无水硫酸镁,规格:AR,南京生健泉化玻仪器有限公司产;异佛尔酮二异氰酸酯(IPDI),纯度>99%,上海麦克林生化科技有限公司产;六亚甲基二异氰酸酯(HDI),纯度>99%,上海麦克林生化科技有限公司产。
仪器包括:磁力加热搅拌器,艾卡仪器设备有限公司产;旋转蒸发仪,上海亚荣生化仪器厂产;低温恒温循环冷却机,西安夏溪电子科技有限公司产;循环水真空泵,巩义市予华仪器有限责任公司产;高真空油泵,威伊真空设备有限公司产;傅里叶变换红外光谱仪,赛默飞世尔有限公司产;核磁共振仪,德国布鲁克科技有限公司产;差示扫描量热(DCS)仪(高温),瑞士-梅特勒托利多公司产;DCS(低温),美国TA仪器公司产;凝胶渗透色谱仪,上海沃特斯智能科技有限公司产。
1.2 合成路线
目前已有的含能粘合剂中,GAP的合成路线经过几十年的研究发展,工艺方法安全、成熟,叠氮化的取代率很高,分子量易于控制,反应的副反应少。因此本文首先尝试进行以GAP作为分子主链进行 3种不同端基PUA的合成试验,合成路线(以GAP/TDI/HEA-PUA为例)如图1所示。图1中,反应(a)为ECH在路易斯酸催化下的阳离子开环聚合反应,生成产物为PECH;反应(b)为PECH和叠氮酸根的亲核取代反应,生成产物为GAP;反应(c)为GAP中羟基进攻异氰酸酯基团的亲核加成反应,生成产物为GAP/TDI/HEA-PUA。
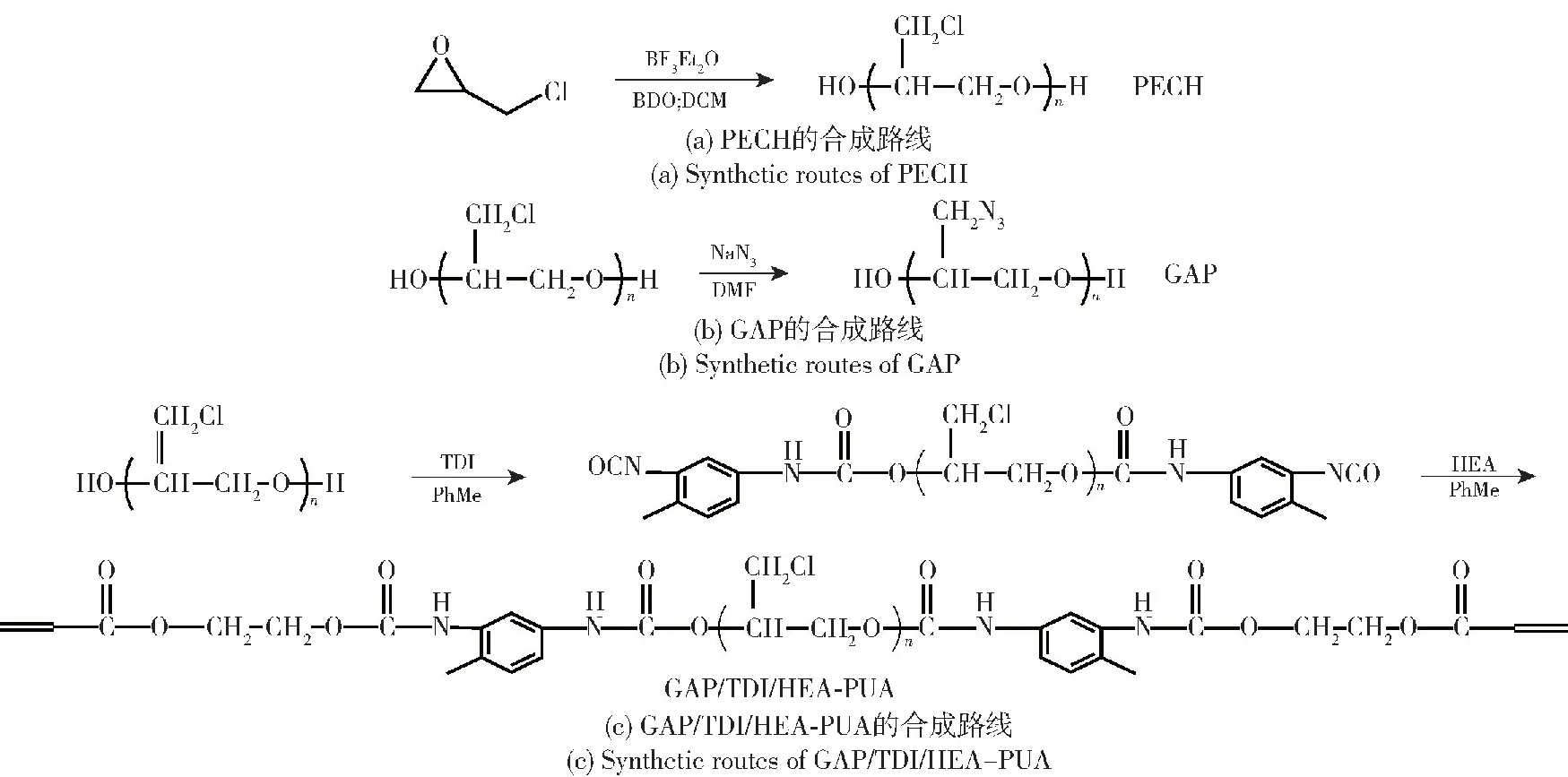
图1 GAP/TDI/HEA-PUA的合成反应路线
1.3 实验
1.3.1 所用试剂的纯化处理和制备方法
1)无水ECH:加入适量氢化钙于120 ℃搅拌回流4~6 h,冷却,使用真空循环水泵在80 ℃~120 ℃下逐步升温减压蒸馏,于试剂瓶内加入4A分子筛封口保存。
2)无水DCM:加入适量氢化钙于50 ℃搅拌回流4~6 h,冷却,使用真空循环水泵在室温下减压蒸馏(使用0 ℃冷浴循环),于试剂瓶内加入4A分子筛封口保存。
3)BDO:加入适量氢化钙于120 ℃加热搅拌2 h,使用高真空油泵逐步升温直至蒸出,于样品瓶内加入4A分子筛封口保存,置于干燥器中。
4)BF3Et2O溶液:加入适量氢化钙,于130 ℃回流2~3 h,冷却,真空循环水泵下90 ℃~120 ℃逐步升温蒸出(勿蒸干),确保产物为无色透明,置于干燥器中封口保存,变黄应重新蒸馏。
5)无水甲苯(PhMe):将甲苯倒入三口圆底烧瓶中,氮气保护下,取少量金属钠(保存在二甲苯中),擦去表面残留的二甲苯,切成细条状加入体系内,待无气泡产生后,加入约等量的二苯甲酮,停止通氮气,体系密闭(冷凝管套气球),于120 ℃搅拌回流至深蓝色(表明体系已除水完全),待冷却,使用真空循环水泵在60 ℃~80 ℃减压蒸馏,得到无水甲苯,转移至试剂瓶内加4A分子筛封口,并置于干燥器中保存。
1.3.2 PECH的合成
氮气保护下,在一个配备有三通玻璃阀门、恒压滴液漏斗、温度计、磁力搅拌器和低温恒温循环冷浴的三口夹套反应器中,加入10 mL无水DCM,再先后加入2.5 mL BDO和2.0 mL BF3Et2O,25 ℃下搅拌络合30 min。络合反应结束后,开启低温冷浴循环,使体系内温度保持在0 ℃左右。氮气氛围下,取92.5 g(1.0 mol)无水ECH与等量的无水DCM混合,转移至恒压滴液漏斗中开始逐滴加入体系,控制在4~6 h滴加完毕,之后继续反应4~6 h。反应结束后用去离子水淬灭反应,洗涤,干燥,过滤后在30 ℃~40 ℃左右旋蒸,除去溶剂DCM,得到产物为无色透明胶体。
1.3.3 GAP的合成方法
取92.5 g PECH溶于等质量的DMF中,转移至圆底烧瓶中,在室温下搅拌过程中加入78 g(1.2 mol)叠氮化钠,升温至100 ℃反应18~24 h,反应过程中体系由白色浑浊变为橘黄色浑浊。反应结束后,洗涤,干燥,过滤后得到有机相。旋蒸除去溶剂DCM和残余的DMF,得到透明橘红色胶体约39 g,得率约为74%。
1.3.4 GAP/TDI/HEA的合成方法
对所制备的GAP根据国家标准GB/T 12008.3—2009测定羟值(OHV),根据羟值进行定量封端反应。GAP在使用前,使用高真空油泵在 80 ℃ 下除水至无气泡产生。氮气保护下,在一个配有玻璃三通阀门、恒压滴液漏斗和磁力加热搅拌器的三口烧瓶中。取3 g GAP(OHV=27.5 mgKOH·g-1)溶于10 mL无水甲苯(PhMe)中,加入约5~10 mg(0.15%~0.30%)催化剂DBDTL,混合均匀后转移至恒压滴液漏斗中。取约0.256 g(1.47×10-3mol)TDI加入圆底烧瓶中,设置温度为60 ℃,开始缓慢滴加GAP-PhMe溶液,控制时间在30 min左右。加料结束后,继续反应30 min,氮气保护下,向体系内加入约9 mg(0.3%)阻聚剂MEHQ。待体系冷却至室温后,取约0.188 g(1.6×10-3mol)HEA缓慢加入体系,控制反应温度维持在室温。加料结束后,体系加热至60 ℃进行反应,当反应液呈浑浊不透明时认定为反应结束。待冷却后停止搅拌,有橘红色不透明胶体析出,将产物用甲苯洗涤1~2次,除去产物中的催化剂和阻聚剂,然后溶于DCM,并加入无水硫酸镁干燥过夜,干燥完毕后过滤得到有机相,旋蒸除去溶剂,得到最终产物橙红色胶体。
1.3.5 GAP/IPDI/HEA的合成方法
合成方法参考1.3.3节,由于IPDI不存在苯环的共轭电子效应,异氰酸酯基团的活性较弱,在保持催化剂等各反应原料用量不变的同时,将反应温度从TDI的60 ℃升高至70 ℃,其余反应条件和方法保持一致。
1.3.6 GAP/HDI/HEA的合成方法
氮气保护下,在三口烧瓶中加入0.841 g(0.005 mol)HDI,取30 mg(0.3 wt.%MEHQ)和0.639 g(0.005 5 mol)HEA溶于10 g无水甲苯并转移至滴液漏斗中,设定反应温度为80 ℃,开始缓慢滴加HEA-PhMe溶液,滴加完毕后继续反应1.5 h。取12 g(0.002 75 mol)GAP(OHV=28.05 mgKOH·g-1)溶于约24 g无水甲苯,在氮气保护下加入反应体系。反应结束后静置溶液出现分层,取下层橙色的胶状产物,加入适量甲苯洗涤1~2次,将产物用DCM溶解后加入无水硫酸镁,干燥过夜。干燥完毕后过滤得到有机相,旋蒸除去溶剂,得到最终产物橙红色胶体。
2 结果与讨论
2.1 表征与测试分析
2.1.1 傅里叶变换红外光谱(FT-IR)
对所合成的中间体化合物PECH进行红外结构表征(见图2)。其中,3 540 cm-1处宽而弱的吸收峰代表端位的羟基;2 940 cm-1、2 880 cm-1为亚甲基和次甲基的C—H伸缩振动峰;1 096 cm-1为C—O—C的不对称伸缩振动峰;738 cm-1为C—Cl的伸缩振动峰。
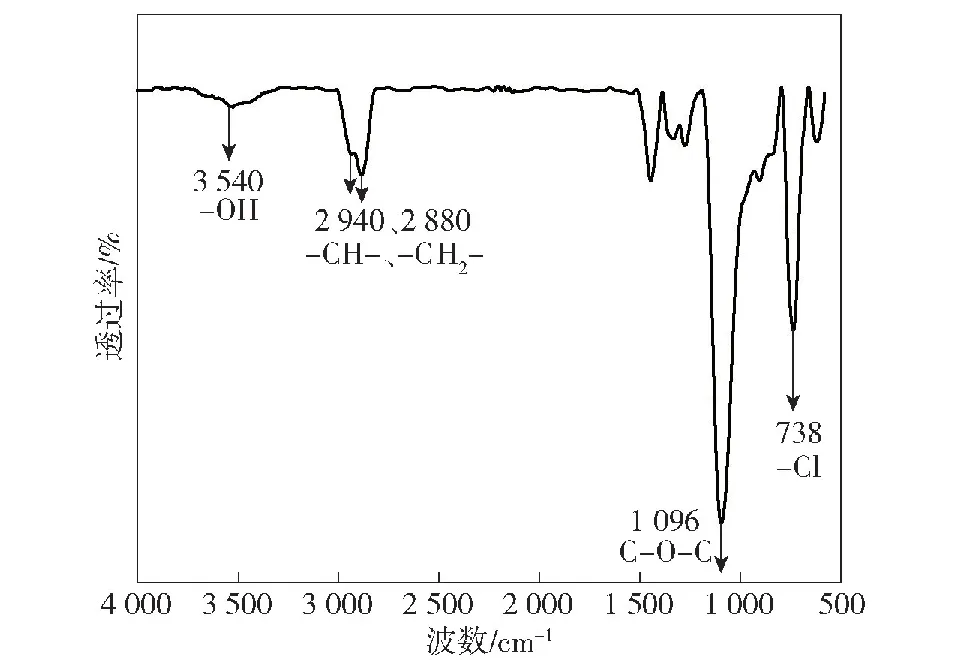
图2 PECH的傅里叶变换红外光谱图
对所合成的GAP进行红外结构表征(见图3)。在750~700 cm-1之间,C—Cl的伸缩振动峰已基本消失。此外,2 090 cm-1为叠氮基团的强吸收特征峰,证明了取代反应进行得十分完全。
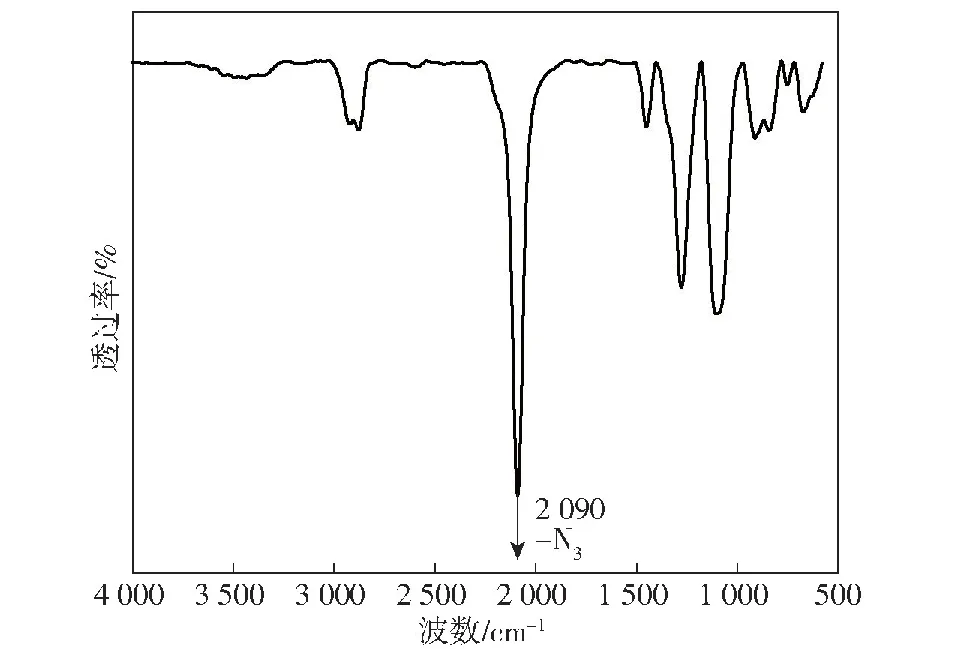
图3 GAP的傅里叶变换红外光谱图

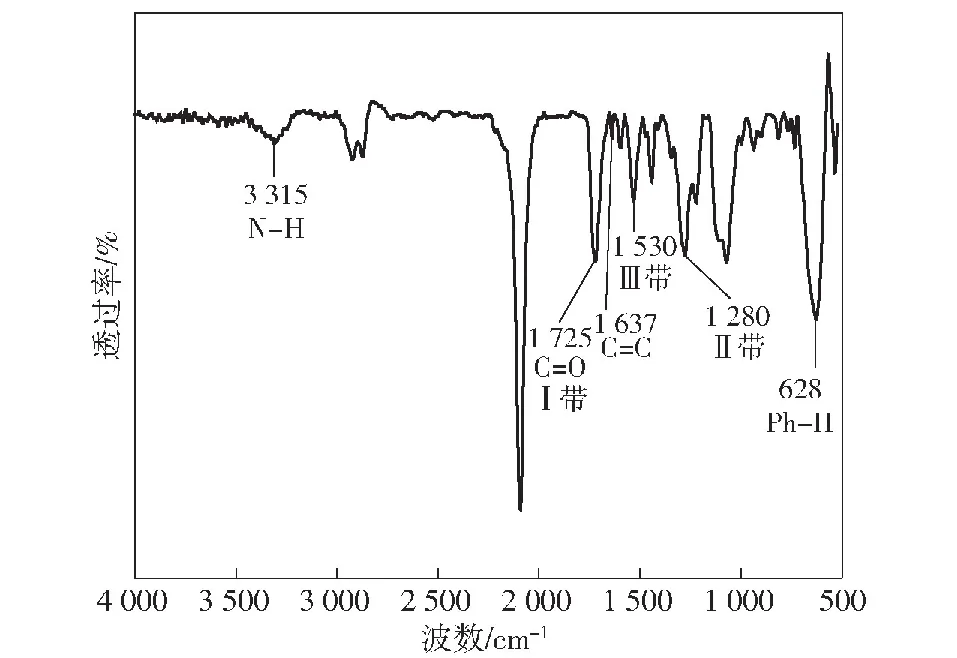
图4 GAP/TDI/HEA-PUA的傅里叶变换红外光谱图
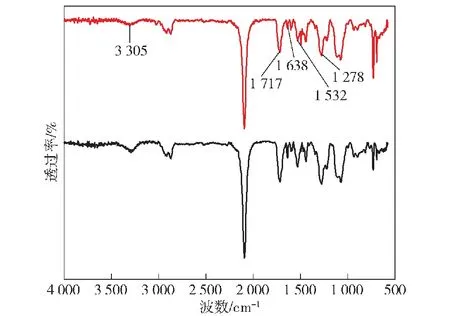
图5 GAP/IPDI/HEA-PUA和GAP/HDI/HEA-PUA的FT-IR谱图
2.1.2 核磁共振氢谱
对所合成的中间体化合物PECH进行核磁共振氢谱表征,结果如图6所示。多重峰分析:1H NMR (500 MHz, DMSO- d6)δ5.74 (s, 1H), 3.79 (d,J=11.5 Hz, 39H), 3.73~3.59 (m, 127H), 3.54 (td,J=11.5, 11.0, 6.7 Hz, 29H), 1.11 (t,J=7.0 Hz, 3H)。
对所合成的中间体化合物GAP进行核磁共振氢谱表征,结果如图7所示。GAP的氢原子所处化学环境与中间体PECH基本一致,化学位移差别不大。多重峰分析:1H NMR (500 MHz, DMSO-d6)δ5.27 (t,J=6.7 Hz, 1H), 3.74~3.54 (m, 54H), 3.46 (q,J=9.9, 7.7 Hz, 29H), 3.33 (q,J=11.9, 8.8 Hz, 25H), 1.57 (d,J=6.3 Hz, 2H)。

图7 GAP的核磁共振氢谱(DMSO)
对所合成的预聚物GAP/TDI/HEA-PUA进行核磁共振氢谱表征,如图8所示。目标产物的主要特征结构为苯环、仲氨基和不对称双键,对应的化学位移值分别为:苯环结构对应于7.17~7.26 mg/L处的多重峰;仲氨基对应于5.04 mg/L处的单峰;不对称双键对应于5.89 mg/L、6.18 mg/L和6.44 mg/L 3处的多重峰。多重峰分析:1H NMR (500 MHz, Chloroform-d)δ7.35~7.12 (m, 15H), 6.46 (dd,J=17.4, 4.0 Hz, 1H), 6.17 (dd,J=17.3, 10.5 Hz, 1H), 5.88 (dd,J=10.4, 3.3 Hz, 1H), 5.05 (s, 2H), 4.56~4.34 (m, 7H), 3.90~3.47 (m, 74H), 3.47~3.17 (m, 42H), 2.24~2.13 (m, 4H), 1.64 (t,J=15.3 Hz, 4H), 1.35~1.13 (m, 3H)。

图8 GAP/TDI/HEA-PUA的核磁共振氢谱(CDCl3)
对所合成的预聚物GAP/IPDI/HEA-PUA进行核磁共振氢谱结构表征,结果如图9所示。多重峰分析:1H NMR (500 MHz, Chloroform-d)δ6.49 (d,J=17.4 Hz, 1H), 6.24~6.16 (m, 1H), 5.92 (d,J=10.2 Hz, 1H), 5.06 (s, 1H), 3.01~2.88 (m, 1H), 1.71 (dd,J=34.6, 18.4 Hz, 5H), 1.31 (d,J=5.5 Hz, 4H), 1.20~1.05 (m, 6H), 0.99 (s, 5H)。

图9 GAP/IPDI/HEA-PUA的核磁共振氢谱(CDCl3)
对所合成的预聚物GAP/HDI/HEA-PUA进行核磁共振氢谱结构表征,结果如图10所示。多重峰分析:1H NMR (500 MHz, Chloroform-d)δ6.51 (d,J=17.4 Hz, 1H), 6.27~6.14 (m, 2H), 5.93 (d,J=11.6 Hz, 1H), 4.47~4.21 (m, 1H), 3.87~3.25 (m, 50H), 2.39 (s, 1H), 1.42 (d,J=76.0, 38.8, 15.6, 8.0 Hz, 1H)。
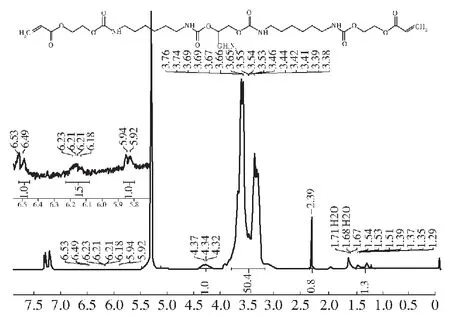
图10 GAP/HDI/HEA-PUA的核磁共振氢谱(CDCl3)
2.1.3 凝胶渗透色谱
对所合成的3种目标预聚物进行GPC测试,对其相对分子质量进行表征,其中Mn为预聚物的数均相对分子质量,Mw为预聚物的重均相对分子质量,多分散性为重均分子量和数均分子量的比值。预聚物中间体GAP的数均相对分子质量为6 561,结合所设计的分子结构通式可以进一步估算出目标预聚物的理论数均相对分子质量(见表1)。
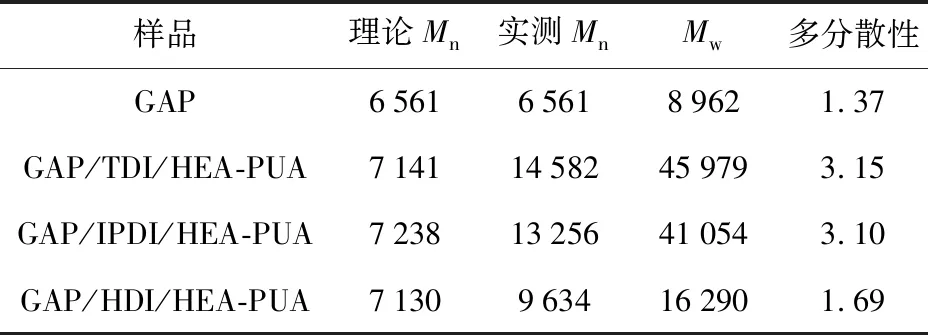
表1 3种目标预聚物的GPC测试结果
由表1可知,3种预聚物的实际相对分子质量均大于理论值,其中GAP/HDI/HEA-PUA相对分子质量的偏差较其他两种预聚物小。这可能是因为TDI和IPDI中的异氰酸酯基团活性较高,且反应过程中由于催化剂的加入,减小了两基团的活性,导致反应的第一阶段发生了一定程度的扩链反应并生成聚氨酯副产物,导致聚合物主链的分子量增大,从而影响了目标预聚物的相对分子质量及其分布。GAP/HDI/HEA-PUA采用了相反的加料顺序,且第一阶段中加入了稍过量的HEA,从而保证异氰酸酯基团反应完全,基本不存在未参与反应的HDI,从而避免了后续过程中与GAP发生反应得到聚氨酯副产物,因此相对分子质量偏差较小。
2.2 引发剂体系用量对PECH数均相对分子质量的影响
通过改变引发剂体系的用量,可以对PECH的相对分子质量进行调控,并通过羟值滴定法计算得到产物的数均相对分子质量,如图11所示。
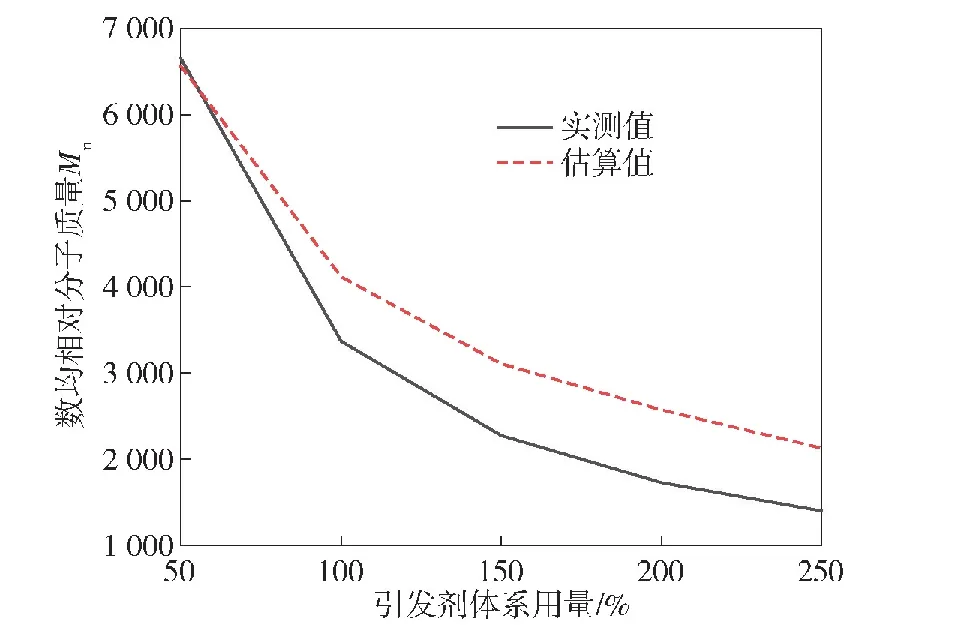
图11 引发剂体系用量对PECH数均相对分子质量的影响
实验所测数据表明,当引发剂含量增加时,产物的理论数均相对分子质量和实际数均相对分子质量均减小。当引发剂含量较低时,可以得到和预期相对分子质量相一致的产物,但随着引发剂用量的增加,产物的实际相对分子质量逐渐小于理论值,且随着分子量的降低,该偏差增大。这可能是因为在不改变单体用量情况下,随着引发剂含量的增加,反应体系中单体的浓度不足,从而导致引发剂或者中间体发生了副反应,造成了在同一分子链上存在多个BDO分子,减少了活性链的实际数量,从而减小了产物的数均相对分子质量。另一方面,当延长反应时间至10 h和12 h时,两次实验产物的产率分别为100%和110%,并且羟值测试的结果也相比正常反应的产物明显偏低,这可能是因为发生了活性中心向溶剂DCM转移的副反应,导致少量DCM作为反应物参与了链聚合反应,使聚合物的数均相对分子质量增大。
2.3 热性能测试
2.3.1 热稳定性测试
使用高温DSC对目标化合物的热稳定性进行研究,并与中间体GAP进行比较,如图12所示。
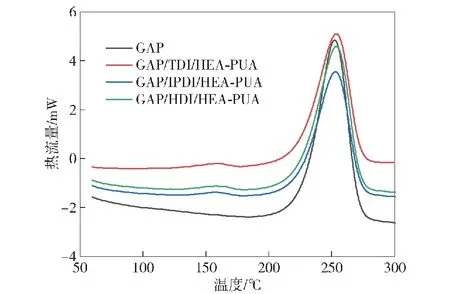
图12 GAP与3种预聚物的高温DSC曲线
GAP/TDI/HEA-PUA的主要放热峰起始温度为224 ℃,终止温度为272 ℃,最高峰温度为250 ℃,与中间体GAP的主要放热峰一致,表示主链结构中叠氮基团的分解放热。除此之外,在140 ℃~170 ℃范围内存在一个微弱的放热峰,相比于主要放热峰热值1 478 J/g,该放热峰热值仅有67 J/g,放热量很小,表示预聚物中双键的热聚合放热,进一步证明目标预聚物结构中存在光敏基团碳碳双键。3种不同端基结构的目标预聚物的DSC放热曲线完全一致,均存在两个放热峰,较小的为双键聚合放热,主峰为叠氮基团分解放热。除此之外,GAP与3种预聚物的热分解温度范围基本相同,表明GAP进行端羟基光固化改性后,聚合物的热分解性能影响较小。
2.3.2 玻璃化转变温度测试
根据图13所示3条DSC曲线,GAP/HDI/HEA-PUA具有最低的玻璃化转变温度为-55.8 ℃,其次分别为GAP/IPDI/HEA-PUA的-52.7 ℃和GAP/TDI/HEA-PUA的-43.8 ℃,表明以脂肪族二异氰酸酯作为原料所制备的PUA预聚物具有良好的分子柔韧性,其中HDI的长链结构相比于脂环结构的IPDI具有更高的运动能力,因此表现出更低的玻璃化转变温度。对于具有刚性的苯环结构的甲苯二异氰酸酯,GAP/TDI/HEA-PUA预聚物表现出较高的玻璃化转变温度,这是因为苯环的存在束缚了分子链的运动能力,随着温度的下降,分子链的运动能力下降更快,从而转变为不具备高弹性的玻璃态。另一方面,3种预聚物的玻璃化转变温度随着实际数均分子量的增加而增加,这是因为聚氨酯在分子量增加的同时,也会生成更多的氨基甲酸酯等基团,基团间会产生氢键,影响分子链柔顺性和力学性能。以上结果表明减小聚合物主链分子长度有利于改善聚合物的低温力学性能。
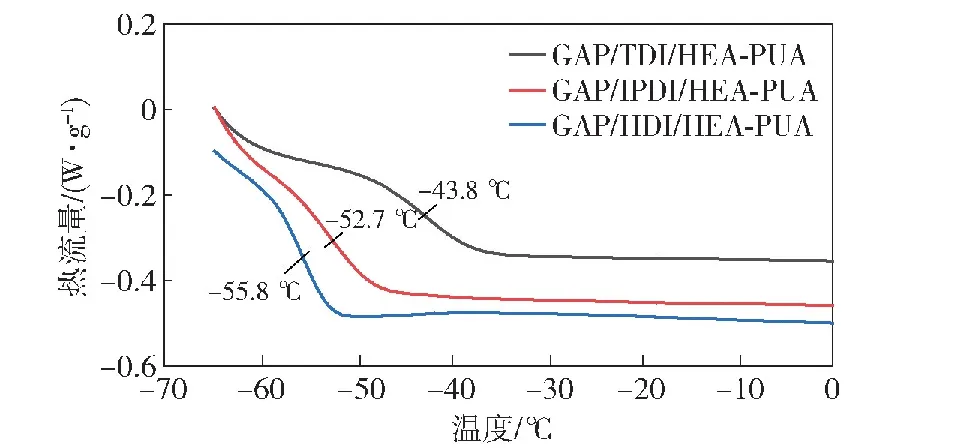
图13 3种预聚物的低温DSC曲线
2.4 固化膜的力学性能测试
将3种预聚物与30%稀释剂HEA以及1%的光引发剂二苯基-(2,4,6-三甲基苯甲酰)氧磷(TPO)复配,制得光敏树脂,将其浇注到哑铃型硅胶模具中,然后放入紫外灯箱中,定时照射2 min,得到光固化膜。
采用拉伸试验机对固化膜的力学性能进行测试,结果如图14所示,3种预聚物组成的光敏树脂固化膜的力学性能表现出较大的差异。其中由GAP/IPDI/HEA-PUA所制备的光固化膜具有最大的断裂延伸率,为470%,同时抗拉强度较高,为0.38 MPa,而由GAP/TDI/HEA-PUA所制备的光固化膜具有最大的抗拉强度,为0.41 MPa,但相比之下断裂延伸率却大幅度降低。而由GAP/HDI/HEA-PUA所制备的光固化膜十分柔软,导致其无论抗拉强度和断裂延伸率均不如其他两种固化膜,这可能是由于HDI的长链结构相比于脂环、苯环结构的二异氰酸酯空间分布更广,电子云密度低,分子间发生交联固化的能力较弱,从而导致在相同的光引发剂含量下,光固化性能较差,导致固化不完全、交联密度较低引起的。

图14 3种目标预聚物的力学性能测试
综合比较,GAP/IPDI/HEA-PUA为预聚物,加入30%的HEA和1%的TPO所制备的光固化膜具有最优异的力学性能,与广泛使用的丁羟胶的力学性能较为接近,满足推进剂对粘合剂的基本力学性能要求。通过减小预聚物分子主链的长度以及增加活性稀释剂的含量和官能度,可以提高光敏树脂材料的双键密度,从而提高光固化的交联密度,对其力学性能进行调节,随着交联密度的增加,一般呈现出抗拉强度升高而断裂延伸率下降的趋势。另一方面,可以通过增大光引发剂用量和提高光辐射强度直接使交联密度增加。此外,还可以将这3种预聚物按不同比例相互混合,制备光敏树脂。因此,可以通过上述5种方法对GAP/IPDI/HEA-PUA光固化膜的力学性质进行调节,最终得到符合预期的光敏树脂配方。
3 结论
1)本文合成了以GAP为预聚物主链的3种光固化含能预聚物GAP/TDI/HEA、GAP/IPDI/HEA和GAP/HDI/HEA。
2)通过高低温DSC分析对3种预聚物的热稳定性和玻璃化转变温度进行测试,预聚物的热聚合起始温度为140 ℃,热分解起始温度为220 ℃,玻璃化转变温度分别为-43.8 ℃、-52.7 ℃和-55.8 ℃,表现出良好的热稳定性和低温力学性能。
3)通过预聚物与活性稀释剂HEA和光引发剂TPO制备3种预聚物的含能光敏树脂材料,进一步制备光固化膜进行力学性能测试,3种光固化膜的抗拉强度在0.1~0.5 MPa之间,断裂延伸率在150%~500%之间。其中GAP/IPDI/HEA-PUA固化膜具有与丁羟胶粘合剂最为接近的力学性能,是一种具有应用前景的新型光固化含能粘合剂。
