一例ISPD基因复合杂合变异所致的肢带型肌营养不良症2U型的诊断和基因检测分析
2023-07-05黄娟缪文华郭晓峰吉炜
黄娟,缪文华,郭晓峰,2,吉炜,3
一例基因复合杂合变异所致的肢带型肌营养不良症2U型的诊断和基因检测分析
黄娟1,缪文华1,郭晓峰1,2,吉炜1,3
1. 福建省儿童医院心内科,福州 350011 2. 福建省妇幼保健院儿科,福州 350001 3. 上海交通大学医学院附属上海儿童医学中心心内科,上海 200127
肢带型肌营养不良症(limb-girdle muscular dystrophy,LGMD)是一组罕见的非先天性遗传性肌肉疾病,主要表现为四肢近端肌张力及肌力进行性减退,其临床表现及遗传模式具有异质性。本研究报道了1例肢带型肌营养不良症2U型10岁男性患者,运动后出现双下肢肌肉无力,入院查肌酸激酶明显升高,经水化碱化治疗后无效。利用高通量测序方法对患者及其父母、妹妹进行肌营养不良相关基因检测,该患者存在基因外显子9杂合缺失以及c.1231C>T(p.Leu411Phe)的杂合错义变异,其父亲携带基因c.1231C>T(p.Leu411Phe)的杂合错义变异,母亲及妹妹携带基因外显子9杂合缺失,上述变异在现有的数据库及文献中均未见报道。对变异位点的保守性分析和蛋白结构预测分析表明,上述位点保守性高,且均位于ISPD蛋白C端结构域,可能对蛋白功能产生影响。综合以上结果并结合相关临床资料,可明确诊断该患者为肢带型肌营养不良症2U型。本文通过总结该患者临床特点及对基因新变异进行分析,丰富了基因变异谱,有助于该病早期诊断及遗传咨询。
肢带型肌营养不良症2U型;基因;基因变异
肢带型肌营养不良症(limb-girdle muscular dystrophy,LGMD)是一组遗传模式及临床表现具有异质性的非先天性遗传性肌肉疾病,发病率约为1/20,000,主要表现为四肢近端(髋/肩带)肌张力减退、肌无力、不同程度的挛缩以及骨骼肌活检营养不良改变等,目前已有报道数百个致病基因变异可导致LGMD[1~3]。基因,也被称为CDP-1-核糖醇焦磷酸化酶 A()基因,位于人类染色体7p21,与α-肌营养不良聚糖(α-dystroglycan,α-DG)糖基化功能有关,该基因发生变异可导致各种类型的肌营养不良症,从轻型LGMD到致命的Walker-Warburg 综合征临床变异型(伴有脑部和眼部受累)[4]。1995年在欧洲神经肌肉中心主持的第一次联盟会议将LGMD根据遗传模式分为两大类:常染色体显性遗传和常染色体隐性遗传[5],此后每报告一个新的染色体基因座,按时间顺序在对应类型下添加一个字母(LGMD1A~LGMD2Z),目前随着外显子组和基因组测序等诸多新技术的更新,LGMD 亚型不断增加,已确定30多种LGMD 亚型[6]。基因变异主要引起的临床亚型为LGMD2U,为常染色体隐形遗传[2]。本文主要报道了1例由基因变异导致的LGMD2U患者,探讨该疾病的临床特点,并报道2个新的变异位点,补充了基因的致病变异数据库,对该类疾病的早期诊断提供参考及借鉴。
1 对象与方法
1.1 对象
患者,男,10岁,汉族,以“运动后双下肢无力7天”为主诉于2022年3月入住我院,父母非近亲结婚,实验室检查提示肌酸激酶明显升高,血清病原学检测结果均为阴性,经水化碱化等治疗后无明显好转,经遗传咨询,家属同意进行遗传学检测和研究。本研究获得了福建省儿童医院人类研究机构委员会的批准(批准伦理号:2022ETKLR12071),患者父母均签署知情同意书。
1.2 全外显子组测序
抽取患者及其父母、妹妹的外周血4~6 mL于EDTA抗凝管中,使用血液DNA提取试剂盒(北京天根生化科技有限公司)的步骤从患者及父母的外周血白细胞中提取总DNA 3 μg的基因组DNA经超声破碎仪(Diagenode,比利时)处理后得到DNA片段,采用KAPA HTP Library Preparation Kit Illumina®Platforms试剂盒(KAPA Biosystems,美国)建库,使用Hybridization capture of DNA libraries using xGen®Lockdown®Probes and Reagents试剂盒(IDT,美国)进行杂交捕获,最后采用Novaseq6000/PE150 (Illumina,美国)进行测序,测序深度100×。
1.3 Sanger测序验证
对患者及其父母、妹妹的DNA标本进行PCR扩增,并对最终变异位点c.1231C>T(p.Leu411Phe)进行Sanger测序验证,所使用的引物序列如下:Forward 5′-GTCATACCGAAGACGGCGAG-3′,Reverse 5′-TGCATCGAGCGGAAATCCAT-3′。
1.4 实时定量多聚核苷酸链式反应(real time quantitative polymerase chain reaction, RT-qPCR)
根据先证者筛选得到候选变异区域,设计合成扩增目的区域所需要的引物序列。缺失区域:Chr7:16255690-16255822。基因引物:Forward 5′-GTGGGTAAGATATGAGAAGCCC-3′,Reverse 5′-TTTCCATTTTCTGACTGGGAGGT-3′。使用赛默飞ABI 7500实时荧光定量PCR系统,对先证者及其父母、妹妹DNA样本进行RT-qPCR检测,使用相对定量法计算相对表达值。
1.5 生物信息学分析
通过NCBI数据库(https://www.ncbi.nlm.nih.gov/)搜索研究中所涉及蛋白的氨基酸序列,使用T-coffee在线服务器(https://tcoffee.crg.eu/)对这些序列进行序列比对,使用ESPript 3.0服务器(https://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi)对这些序列进行进一步序列分析。基于human isoprenoid synthase domain-containing protein的晶体结构(PDB ID: 4CVH),使用SWISS-MODEL在线服务器(https://swissmodel.expasy.org/)对目标蛋白和变异体蛋白进行蛋白模型构建,使用PyMOL对这些蛋白模型进行亲疏水性、表面静电势能和蛋白结构分析和作图。
2 结果与分析
2.1 患者临床资料
患者,男,10岁,因“双下肢肌无力7天,发现肌酶升高5天”于福建省儿童医院心血管内科就诊。患者入院7天前运动后出现双下肢无力,无肌肉酸痛、茶色尿等,能直立行走,但易疲乏。否认药物、毒物服用史。既往体健,生长发育史正常。
患者系第一胎第一产,足月顺产,1岁1月独走,智力发育无异常。父母体健,否认近亲结婚,妹妹4岁,体健。体格检查:神志清楚,精神反应可,面色红润,瞳孔等大等圆,对光反射灵敏,心肺腹部查体阴性,四肢肌张力稍减低,双上肢上抬有力,肌力5级,无抬臂和梳头困难,双下肢近端肌力3级,远端肌力4级,上下楼正常,无蹲起、跑步困难等,骨盆带及肩胛骨带肌肉无明显萎缩,双侧膝反射减弱,椎体束征阴性,Gower征阳性,病理征阴性。实验室检查:磷酸肌酸激酶(,CK)4386 U/L(正常参考值:25~200 U/L),肌酸激酶同工酶(CK-MB)54.77 U/L(正常参考值:0~ 25 U/L);血清病原学均阴性;心电图提示一度房室传导阻滞;心脏彩超提示心内结构及心功能未见异常;腹部彩超提示肝大(肋下3.5 cm,右肝斜径12.0 cm),肝实质回声稍增粗,脾稍大;肌电图检查未见明显异常;眼底及颅脑检查均未见明显异常。
入院考虑诊断:肌酸激酶增高待查,可能包括以下方面:(1)肌病性高CK血症,主要包括炎症性肌病、肌营养不良、先天性肌病、内分泌性肌病及代谢性肌病等;(2)非肌病性高CK血症病因,主要包括心肌坏死或受损、肌肉受损或直接缺血、持续高烧不退以及中毒等。予以水化碱化等对症处理后患者CK未见明显下降(4713 U/L),双下肢肌无力也无明显改善。出院后规律随访2月,患者CK波动于4500~5000 U/L,双下肢肌无力较前相仿,尚能直立行走。根据患者临床表现、实验室检查结果及治疗效果,考虑肌营养不良或先天性肌病可能性较大,为进一步明确病因,根据患者家属意愿要求完善全外显子基因检测。
2.2 全外显子组测序和Sanger测序结果
该患者存在基因外显子9杂合缺失以及c.1231C>T(p.Leu411Phe)的杂合错义变异,上述变异在现有的数据库中均未见报道。根据美国医学遗传学学会(American College of Medical Genetics,ACMG)的判定标准[7],基因外显子9杂合缺失为可能致病性(lpathogenic,LP)的基因变异,证据包括:(1)非常强烈的证据(pathogenic-verystrong,PVS1):单个外显子缺失;(2)中等证据(pathogenic-moderate,PM2):检出变异在多个正常对照人群数据库未发现(或隐性遗传病中极低频位点)。序列分析显示基因c.1231C>T变异导致氨基酸出现错义变异(p.Leu411Phe),位置411的亮氨酸由苯丙氨酸替代,根据ACMG判定标准[5],该变异位点为意义不明确(US)的基因变异,其证据包括:中等证据(pathogenic-moderate,PM2+PM3):检出变异在多个正常对照人群数据库未发现(或隐性遗传病中极低频位点);在隐性遗传病中,在受累个体待分类变异反式位置上检测到致病或可能致病变异。
患者父亲携带基因c.1231C>T(p.Leu411Phe)的杂合错义变异,患者母亲及妹妹携带基因外显子9杂合缺失,说明患者该变异分别遗传自父母,符合常染色体隐性遗传模式(图1,A~D)。
2.3 RT-qPCR验证结果
先证者、先证者之父、先证者之母、先证者之妹,定量PCR实验结果中2–ΔΔCt值分别为 0.52、1.02、0.52、0.58,即先证者、先证者之母、先证者之妹基因外显子9杂合缺失,先证者之父基因外显子9正常(图2)。
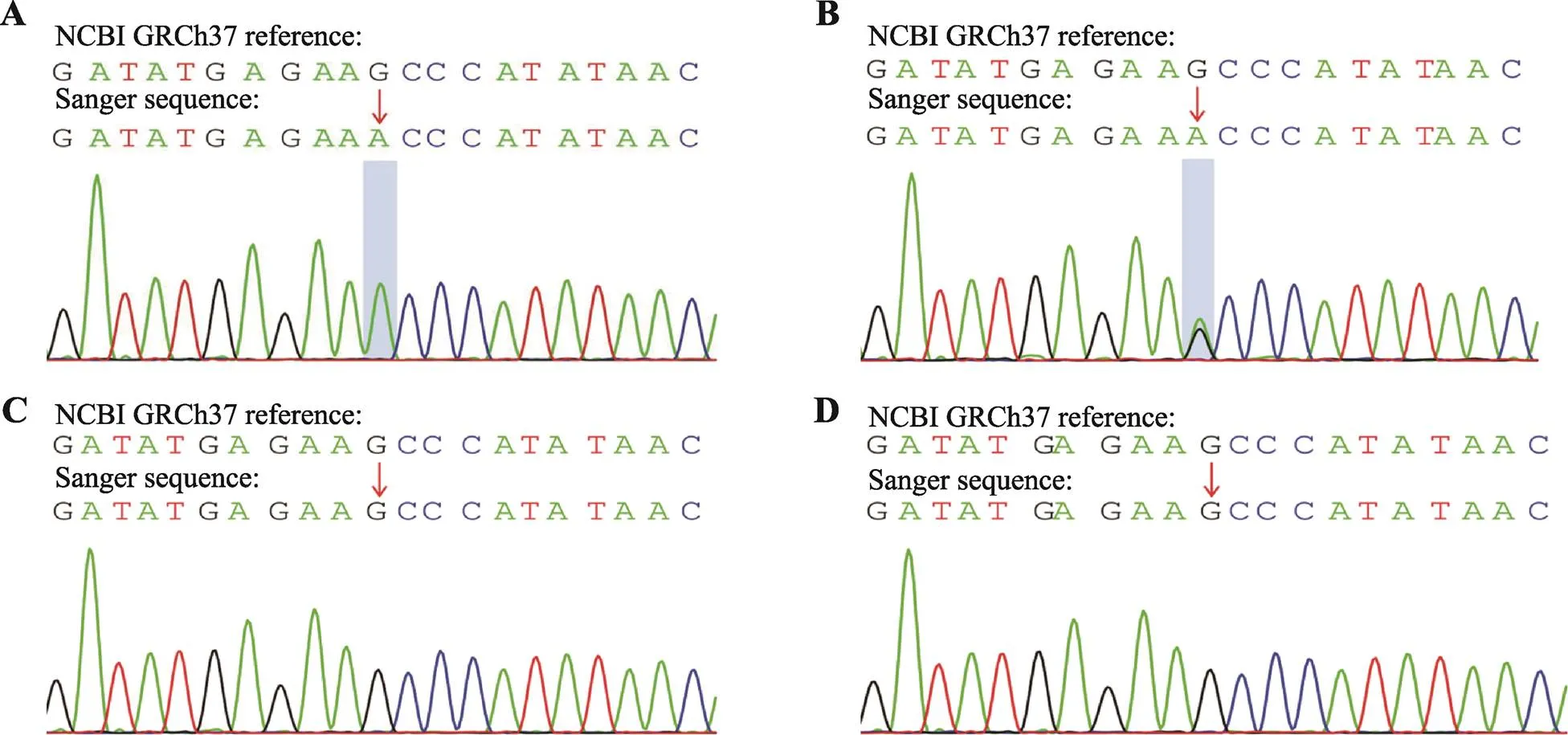
图1 Sanger测序结果
A:先证者(c.1231C>T变异);B:先证者父亲(携带者);C:先证者母亲(野生型);D:先证者妹妹(野生型)。
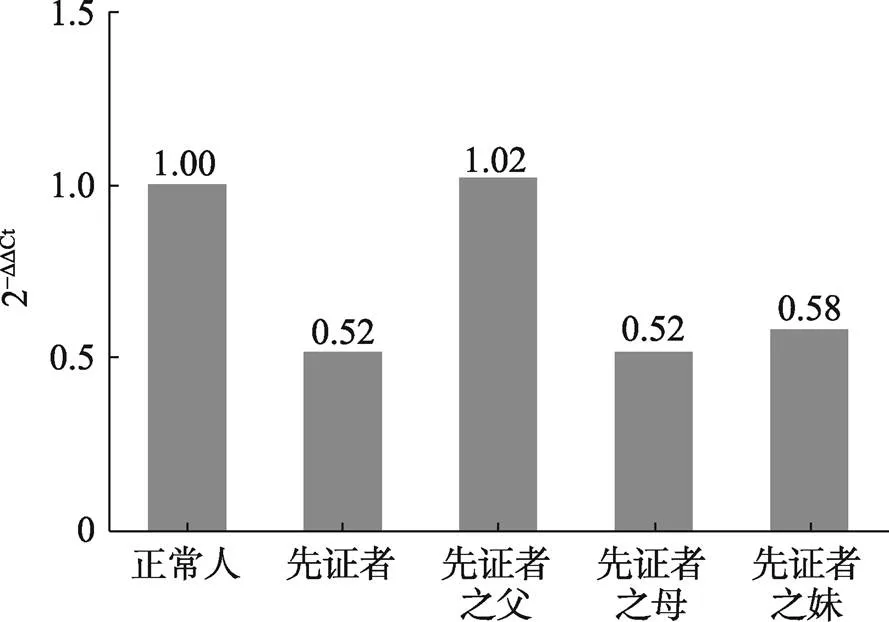
图2 先证者及其家庭成员ISPD基因外显子9杂合缺失RT-qPCR验证结果
2–ΔΔCt<0.1 为纯合缺失,0.3<2–ΔΔCt<0.7 为杂合缺失,0.7< 2–ΔΔCt<1.3为正常,1.3<2–ΔΔCt<1.8为单倍重复。
2.4 变异位点保守性和蛋白结构分析
经过T-coffee在线服务器对人()、恒河猴()、印度野牛()、家马()、穴兔()、小鼠()、斑马鱼()、热带爪蟾()等物种的ISPD蛋白进行保守性分析,发现c.1231C>T变异对应第Leu411氨基酸在多物种中高度保守(图3A),第374~417氨基酸在多物种中保守性较高(图3B),提示该区域氨基酸改变可能对该蛋白的功能产生一定的影响。根据计算机软件预测蛋白结构,发现基因外显子9缺失变异体破坏了C端结构域之间的相互作用,直接影响蛋白完整性及酶活性(图4,A~F),c.1231C>T变异亦位于ISPD蛋白C端结构域(图4,G和H),可能对蛋白功能产生影响。
3 讨论
肢带型肌营养不良症为第四大最常见的遗传肌肉性疾病,其特征为进行性的近端肢带肌肉无力。该病的发病年龄从儿童早期(非先天性)到成年晚期不等,肌无力的进展具有个体差异性以及遗传异质性,儿童临床表现严峻、进展快速而成人表现为相对良性的轻型肢带型肌营养不良症[1,6]。本文分享的病例是1例由基因变异导致的肢带型肌营养不良症患者,结合遗传检测结果,将其临床表型诊断为LGMD2U[2,8]。
基因位于人染色体7p21,编码含有类异戊二烯合酶结构域的蛋白质,证实与α-DG的O-甘露糖基化的初始步骤有关[4]。α-DG是肌营养不良蛋白糖蛋白复合物的高度糖基化核心成分,在肌膜和细胞外基质之间形成联系,参与细胞识别、信号传导等多种生物学过程[9]。肌营养不良蛋白聚糖由两个亚基α和β组成,它们在翻译后被切割以产生高度O-糖基化跨膜蛋白(β)和外膜蛋白(α),而O-甘露糖基化为该糖基化过程的一个重要特殊组成部分,参与α-DG与细胞外基质蛋白(如层粘连蛋白、集聚蛋白)的相互作用[10]。基因的变异会干扰上述生物化学过程,导致肌肉萎缩和无力等症状。该基因的变异首先在最严重的肌营养不良、Walker-Warburg综合征和肌眼脑疾病病例谱中被发现,随着检测技术的进步,逐渐在英国、土耳其、意大利的儿童肌营养不良症队列中发现,结合相关病例临床表型,由基因变异引起的LGMD列入为LGMD2U亚型[2]。LGMD2U亚型可于儿童早期发病,病情进展缓慢,表现为四肢肌张力降低,步态异常及Gowers征阳性等,通常在12岁以内就丧失行走能力,血肌酸激酶可波动于700~9000 U/L[8]。目前,基因变异关联LGMD表型的患者仍较少,尚未在LGMD队列中估计基因变异的患病率。
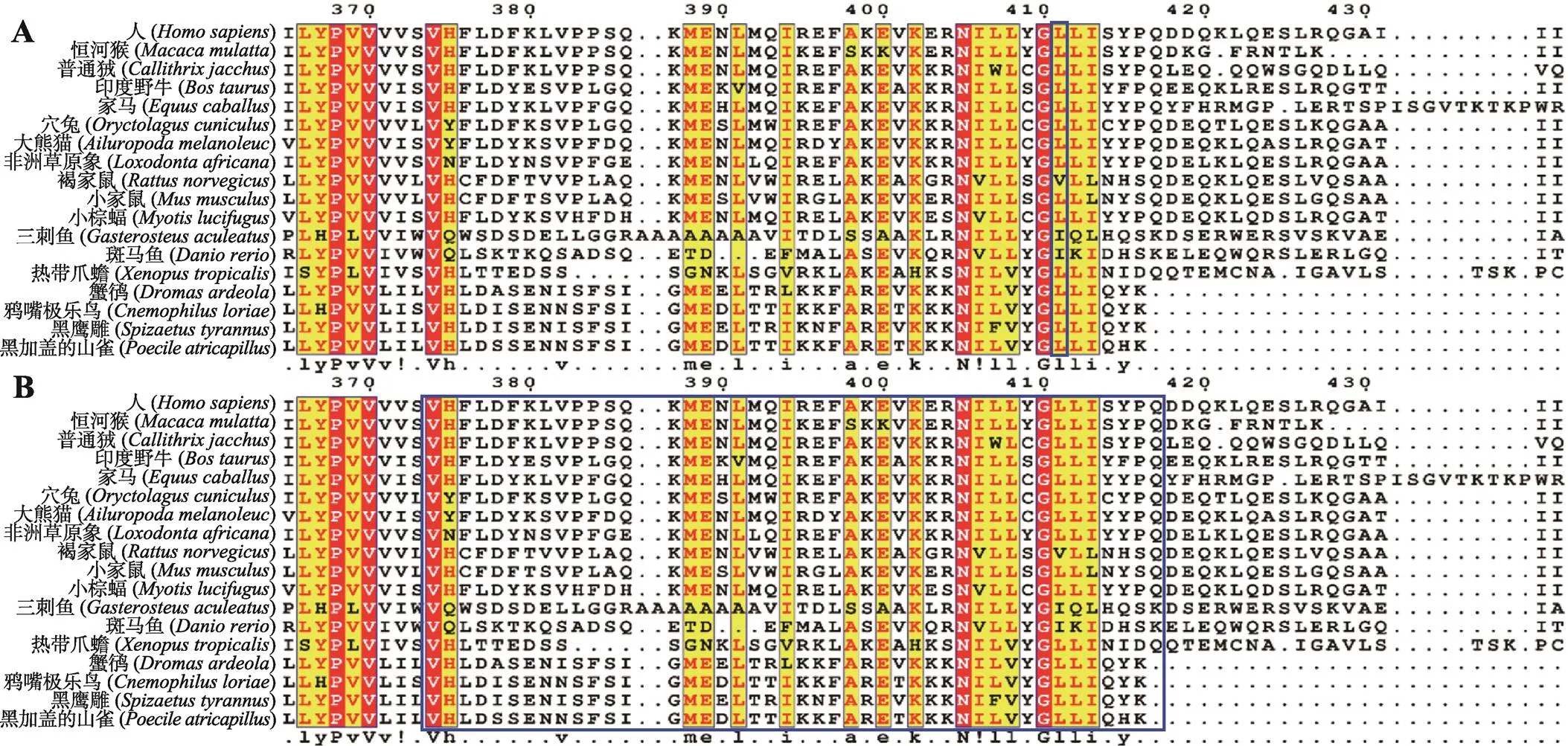
图3 ISPD蛋白变异位点保守性分析
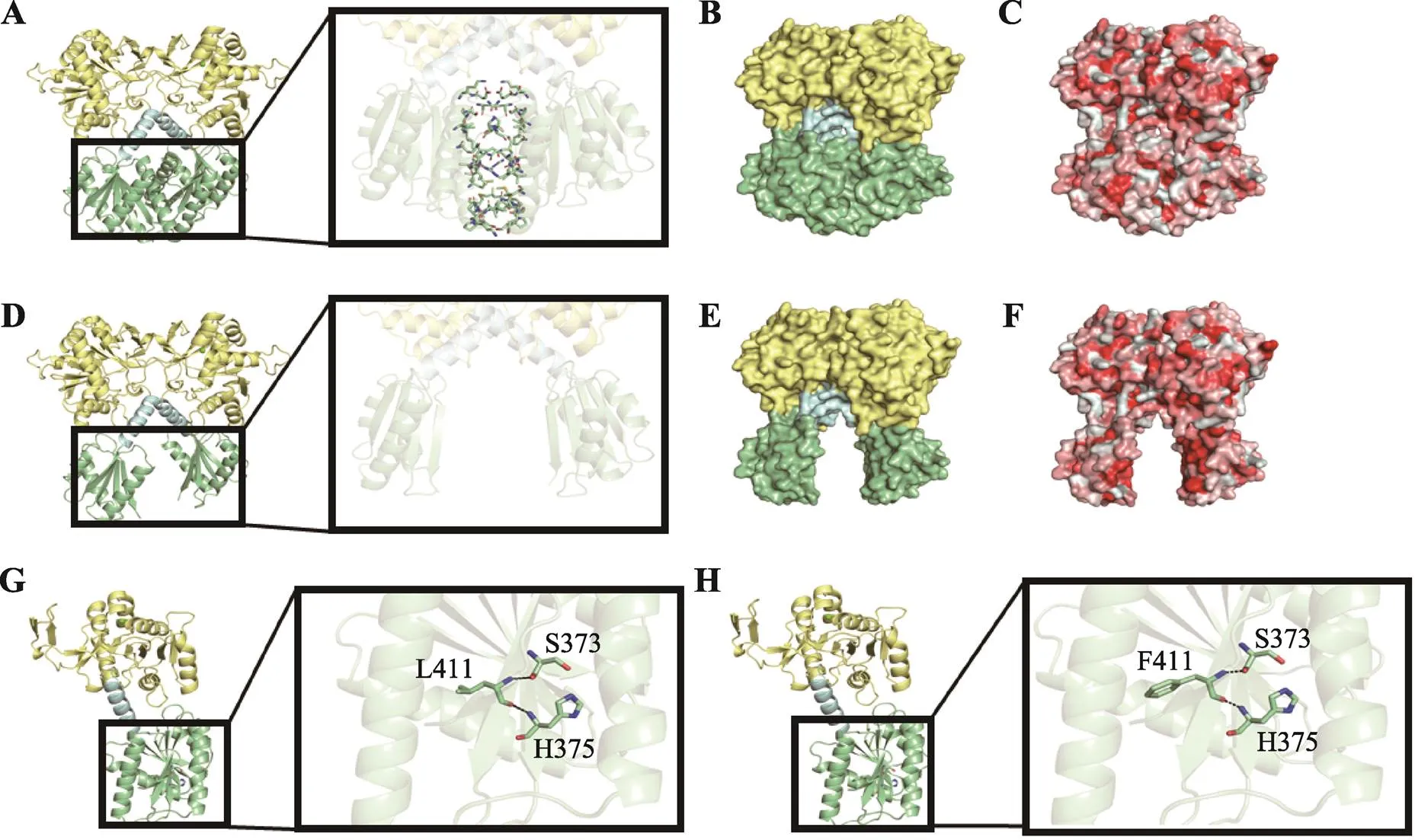
图4 ISPD蛋白结构模拟和预测分析
A:野生型ISPD蛋白结构预测(二聚体);B:野生型静电势能图;C:野生型疏水性分析图(红色疏水,白色亲水);D:外显子9缺失型ISPD蛋白结构预测;E:外显子9缺失型静电势能图;F:外显子9缺失型疏水性分析图(红色疏水,白色亲水);G:野生型ISPD蛋白结构预测(单体);H:c.1231C>T变异型ISPD蛋白结构预测。A、B、D、E、G及H中黄色为N端胞苷酰转移酶结构域(CT domain),蓝色为接头螺旋,绿色为C端结构域(C-terminal domain)。
目前针对LGMD的治疗,虽然转化研究正在不断进行,但除了被归类为LGMD2V的庞贝病有酶替代疗法外,对其他类型的LGMD患者仍是致力于对症治疗并努力预防/解决并发症[3]。有病例报道指出,低剂量泼尼松治疗可改善患者运动功能、延长步行时间并减少摔跤频率[11],但缺乏循证学依据的支持。细胞培养和动物模型有助于阐明发病机制,有可能成为制定治疗方案的关键。近年来,随着研究的深入,分子治疗和基因治疗亦展示出新的治疗潜力。例如,Turan等[12]利用患者衍生的诱导多能干细胞纠正了与LGMD2B相关的基因无义变异c.5713C>T(p.R1905X)和与LGMD2D相关的α-肌聚糖基因错义变异c.229C>T(p.R77C)。此外,还有团队致力于α-肌聚糖缺乏症(LGMD2D)的基因治疗及分子治疗,旨在修复人体肌聚糖复合物的功能,已在体外实验中取得一定成效[13,14]。
本文报道了1例10岁男性LGMD2U患者,该患者在运动后表现出双下肢无力,并伴有明显的CK水平升高,目前可独立行走,无脑部及眼部异常证据。全外显子组测序揭示了该患者存在基因外显子9杂合缺失以及c.1231C>T(p.Leu411Phe)的杂合错义变异。ISPD蛋白具有一个典型的N端胞苷酰转移酶结构域(CT domain),通过一个接头螺旋连接到同源性结构域(C-terminal domain),胞苷酰转移酶结构域采用核苷酸糖焦磷酸酶之间的特征性罗斯曼样α/β折叠形成同源二聚体(图4)。ISPD蛋白的C端结构域除了增强N端胞苷酰转移酶结构域的稳定性、活性和完整性外,还可能具有酶活性,因此C端结构域的截断变异可能导致ISPD蛋白功能丧失[15]。本例患者的外显子9的缺失及第Leu411氨基酸变异均位于ISPD蛋白C端结构域,可能影响蛋白质功能。虽然该患者目前只表现为双下肢无力,但由于LGMD在儿童成长阶段的临床表现呈现高度异质性,该疾病相关的临床表型可能逐渐显现,该患者目前于门诊密切随诊观察,必要时进行特殊治疗。
此外,本例的LGMD2U是一种常染色体隐性遗传疾病,患者的父母都是健康的隐性携带者。如果患者的父母再次怀孕,胎儿有25%概率患上相同的疾病。因此,建议家属在再次妊娠前接受遗传咨询,必要时完善胎儿遗传学检测。本文通过报道1例LGMD2U患者,对其基因外显子9杂合缺失以及c.1231C>T(p.Leu411Phe)的杂合错义变异进行分析,完善基因的致病变异数据库,并为该类疾病的早期诊断和干预提供了重要的临床参考价值。
[1] Wicklund MP, Kissel JT. The limb-girdle muscular dystrophies., 2014, 32(3): 729–749.
[2] Liewluck T, Milone M. Untangling the complexity of limb-girdle muscular dystrophies.2018, 58(2): 167–177.
[3] Chu M L, Ellen M. The limb-girdle muscular dystrophies: is treatment on the horizon?, 2018, 15(4): 849–862.
[4] Cirak S, Foley AR, Herrmann R, Willer T, Yau S, Stevens E, Torelli S, Brodd L, Kamynina A, Vondracek P, Roper H, Longman C, Korinthenberg R, Marrosu G, Nürnberg P, Consortium U, Michele DE, Plagnol V, Hurles M, Moore SA, Sewry CA, Campbell KP, Voit T, Muntoni F.gene mutations are a common cause of congenital and limb-girdle muscular dystrophies.2013, 136(Pt 1): 269–281.
[5] Bushby KM. Diagnostic criteria for the limb-girdle muscular dystrophies: report of the ENMC consortium on limb-girdle dystrophies., 1995, 5(1): 71–74.
[6] Angelini C. LGMD. Identification, description and classification., 2020, 39(4): 207–217.
[7] Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology., 2015, 17(5): 405–424.
[8] Luo SS, Lu JH. Limb-girdle muscular dystrophy., 2019, 52(7): 573–581.罗苏珊, 卢家红. 肢带型肌营养不良. 中华神经科杂志, 2019, 52(7): 573–581.
[9] Muntoni F, Brockington M, Blake DJ, Torelli S, Brown SC. Defective glycosylation in muscular dystrophy., 2002, 360(9343): 1419–1421.
[10] Waite A, Brown SC, Blake DJ. The dystrophin-glycoprotein complex in brain development and disease., 2012, 35(8): 487–496.
[11] Yang H Y, Cai F, Liao H G, Gan S Y, Xiao T, Wu L W. Case report: ISPD gene mutation leads to dystroglycanopathies: genotype phenotype analysis and treatment exploration., 2021, 9: 710553.
[12] Turan S, Farruggio AP, Srifa W, Day JW, Calos MP. Precise correction of disease mutations in induced pluripotent stem cells derived from patients with limb girdle muscular dystrophy., 2016, 24(4): 685–696.
[13] Pedemonte N, Lukacs GL, Kai D, Caci E, Zegarra-Moran O, Galietta LJV, Verkman AS. Small-molecule correctors of defective ΔF508-CFTR cellular processing identified by high-throughput screening., 2005, 115(9): 2564.
[14] Carotti M, Marsolier J, Soardi M, Bianchini E, Gomiero C, Fecchio C, Henriques SF, Betto R, Roberta Sacchetto R, Richard L, Sandonà D. Repairing folding-defective a-sarcoglycan mutants by CFTR correctors, a potential therapy for limb-girdle muscular dystrophy 2D., 2018, 27(6): 969–984.
[15] Riemersma M, Froese DS, van Tol W, Engelke UF, Kopec J, van Scherpenzeel M, Ashikov A, Krojer T, von Delft F, Tessari M, Buczkowska A, Swiezewska E, Jae LT, Brummelkamp TR, Manya H, Endo T, van Bokhoven H, Yue WW, Lefeber DJ. Human ISPD is a cytidyltransferase required for dystroglycan O-mannosylation., 2015, 22(12): 1643–1652.
Diagnosis and genetic testing analysis of limb-girdle muscular dystrophy type 2U caused by a compound heterozygous mutation in thegene
Juan Huang1, Wenhua Miao1, Xiaofeng Guo1,2, Wei Ji1,3
Limb-girdle muscular dystrophy (LGMD), a rare group of non-congenital inherited muscle diseases, is characterized by a progressive reduction in muscle tone and force of the proximal limbs. The clinical manifestations and genetic patterns of LGMD are heterogeneous. This study reported on a 10-year-old male patient with LGMD type 2U who experienced muscle weakness in the lower limbs after exercise. Upon admission, the patient's creatine kinase levels were significantly elevated, and hydration and alkalinization therapy were ineffective. Using high-throughput sequencing, muscular dystrophy-related genes were tested in the patient, his parents, and his sister. The patient was found to have a heterozygous deletion of exon 9 of thegene and a heterozygous missense mutation c.1231C>T (p.Leu411Phe). The patient's father carried the heterozygous missense mutation c.1231C>T (p.Leu411Phe) of thegene, while his mother and sister carried a heterozygous deletion of exon 9 of thegene. These mutations have not been reported in existing databases or literature. Conservation and protein structure prediction analyses of the mutation sites indicated that they are highly conserved and located in the C-terminal domain of the ISPD protein, which may affect protein function. Based on the above results and relevant clinical data, the patient was definitively diagnosed with LGMD type 2U. This study enriched the spectrum ofgene mutations by summarizing the patient's clinical characteristics and analyzing newgene variations. This can aid in the early diagnosis and genetic counseling of the disease.
limb-girdle muscular dystrophy type 2U;gene; gene mutation
2023-02-15;
2023-05-10;
2023-05-19
福建省自然科学基金面上项目(编号:2022J011072)资助[Supported by General Program of Natural Science Foundation of Fujian (No. 2022J011072)]
黄娟,硕士研究生,住院医师,研究方向:临床医学(心血管)。E-mail: 451294832@qq.com
缪文华,硕士研究生,住院医师,研究方向:临床儿科学(心血管)。E-mail: 36163193@qq.com
黄娟和缪文华并列第一作者。
吉炜,博士,副主任医师,研究方向:儿童心血管方向。E-mail: jidou@126. Com
郭晓峰,本科,副主任医师,研究方向:儿科学(心血管)。E-mail: 1637943000@qq. com
10.16288/j.yczz.22-329
(责任编委: 周红文)
