TRIP13基因新突变导致卵母细胞成熟阻滞为特征的女性不孕
2023-07-05吕香江郭静林戈
吕香江,郭静,林戈,
基因新突变导致卵母细胞成熟阻滞为特征的女性不孕
吕香江1,郭静2,林戈1,2
1. 中南大学基础医学院生殖与干细胞工程研究所,长沙 410078 2. 中信湘雅生殖与遗传专科医院,长沙 410008
卵母细胞成熟阻滞(oocyte maturation arrest,OMA)是指卵母细胞减数分裂过程异常而导致的卵母细胞成熟障碍的一种罕见的临床现象,也是女性原发性不孕的原因之一。这类患者临床上常表现为:反复促排卵后无法获得成熟卵子,并且未成熟卵母细胞经体外培养后仍不能成熟。迄今研究发现、和基因突变与OMA有关,但是有关OMA的遗传学分子因素及机制研究仍不完整。本研究通过收集临床上辅助生殖助孕过程中35名反复出现OMA的原发不孕女性患者的外周血,提取其基因组DNA进行全外显子组测序分析,对疑似致病突变进行验证及家系共分离分析,发现先证者1的基因9号外显子存在纯合突变c.859A>G,突变导致对应编码的287位的氨基酸由异亮氨酸突变为缬氨酸(p.Ile287Val);先证者2的基因1号外显子存在纯合突变c.77A>G,突变导致对应编码的26位的氨基酸由组氨酸突变为精氨酸(p.His26Arg);先证者3的基因的4号和12号外显子存在复合杂合突变c.409G>A和c.1150A>G,c.409G>A突变导致对应编码的137位的氨基酸由天冬氨酸突变为天冬酰胺(p.Asp137Asn),c.1150A>G突变导致对应编码的384位的氨基酸由丝氨酸突变为甘氨酸(p.Ser384Gly),三名患者所携带的突变均引起基因编码的TRIP13蛋白发生错义突变。其中3个突变位点在国内外尚未有文献报道。此外,通过在HeLa细胞中分别转染四个突变质粒,并进行体外免疫印记实验和细胞增殖实验,发现这些突变会造成不同程度的TRIP13蛋白表达的增加以及细胞增殖速率异常。本文总结了既往研究报道的基因突变位点,丰富了致病基因突变谱,为进一步研究基因导致OMA的致病机制及临床的诊断和治疗提供了依据。
卵母细胞成熟阻滞;;减数分裂;女性不孕
卵母细胞成熟阻滞(oocyte maturation arrest,OMA)是女性不孕的一种常见病因,患者因卵母细胞减数分裂过程异常导致卵母细胞停留在GV期(germinal vesicle)或MI期(metaphase I)而不能形成成熟卵子[1]。在临床辅助生殖技术中,体外成熟(maturation,IVM)技术可以在体外模拟体内卵母细胞成熟环境,将未成熟的卵母细胞体外培养至MII期(metaphase II),移植后使患者获得成功妊娠[2],但仍有很多患者无法通过辅助生殖技术解决不明原因的卵子不熟问题。
本文报道了3例因卵母细胞成熟阻滞导致不孕的患者,通过全外显子组测序技术发现她们都携带基因突变,患者1携带基因纯合错义突变c.859A>G(p.Ile287Val),患者2携带基因纯合错义突变c.77A>G(p.His26Arg),患者3携带基因复合杂合错义突变c.409G>A (p.Asp137Asn)和c.1150A>G(p.Ser384Gly)。通过对HeLa细胞转染突变质粒进行体外功能实验,结果发现这些突变均导致HeLa细胞中TRIP13蛋白表达增加,此外,进一步细胞增殖实验表明突变使得HeLa细胞增殖速率出现了不同程度的异常。最后,本文总结了既往文献报道的基因突变位点和临床表型谱。
1 对象与方法
1.1 对象及临床资料收集
患者1,女性,28岁,原发不孕8年。患者2,女性,30岁,原发不孕4年。患者3,女性,29岁,原发不孕2年。患者均就诊于中信湘雅生殖与遗传专科医院。在获得中信湘雅生殖与遗传专科医院伦理委员会批准(批准伦理号:LL-SC-2018-009)后,收集患者的病史、促排方案、助孕周期与结局等临床资料;同时经患者及家属知情同意后,收集患者及亲属外周血。
1.2 全外显子组测序及家系验证
收集患者及亲属外周血,使用QIAamp DNA Blood Mini Kit (Qiagen,德国)提取基因组DNA样本。将患者外周血中提取的基因组DNA送至武汉希望组生物科技有限公司进行全外显子组测序分析。利用高通量测序平台进行深度测序,获得100×的测序reads。将reads进行基因组匹配、抠取变异、过滤去除低质量的变异位点,保留高质量的变异位点。将高质量的变异位点与对照人群中的变异位点进行比较,筛选在对照人群中没有或罕见的变异位点;再结合变异位点所在序列的保守性和突变有害性预测,对可疑突变位点进行优先级排序。针对排序靠前的变异,再进行Sanger验证。如果Sanger测序结果与全外显子组测序结果一致,将该变异作为候选致病突变进行下一步分析。
1.3 突变位点的致病性与蛋白质结构预测
通过gnomAD数据库对突变位点发生的频率进行预测,使用REVEL软件,Polyphen2、Mutation Taster工具对突变位点的致病性进行预测;根据美国医学遗传学与基因组学学会(American College of Medical Genetics,ACMG)发布的最新版基因变异解读标准和指南将检测到的变异分类为“致病性变异”、“疑似致病性变异”、“临床意义未明变异”、“疑似良性变异”或“良性变异”。使用UGENE软件对不同物种基因突变位点所在序列进行保守性分析。在NCBI数据库下载TRIP13蛋白的氨基酸序列,将四个突变位点的氨基酸分别改成突变后的氨基酸,利用SWISS-MODEL蛋白质结构建模工具预测野生型(wild-type,WT)和四个突变蛋白的三级结构,选择同一模板且建模质量高的的预测结构,然后使用PyMol软件对野生型和突变型蛋白结构进行比较。本文选择5vqa.1.A模板,覆盖序列大于99.5%,相似度高;通过GMQE(global model quality estimate)值和QMEANDisCo Global值评估预测模型质量,数值越接近1建模质量越好,本文预测的蛋白结构GMQE值为0.8,QMEANDisCo Global值为0.76±0.05,为较高质量预测模型。
1.4 构建突变质粒
野生型质粒由山东维真生物科技有限公司构建,将基因全长编码序列克隆到带有FLAG标签的pENTER载体中。通过SnapGene软件设计构建4种突变质粒的双向引物,使用Mut Express II快速突变试剂盒(Vazyme,南京)分别向野生型质粒中引入4个突变(c.77A>G,c.409G>A,c.859A>G,c.1150A>G)。按照突变试剂盒说明书所示反应体系和反应条件对野生型质粒进行扩增,扩增后去除原始模板并重组质粒,将重组产物转化后进行测序验证,得到4种突变质粒。
1.5 细胞转染
将HeLa细胞系培养于含10%胎牛血清的DMEM (Invitrogen,美国)培养基中,并放置于37℃、5% CO2的培养箱中培养。
使用GeneTwinTM基因转染试剂(Biomed,北京)将野生型质粒和4个突变质粒转染到HeLa细胞中。具体步骤如下:在6孔板的每个孔中各接种1~2×105个HeLa细胞,24 h后(细胞密度在70%~90%时),将2 μg质粒DNA和50 μL DMEM培养基混匀制成DNA稀释液,并将6 μL转染试剂GeneTwinTM和44 μL opti-MEM (Invitrogen,美国)混匀制成GeneTwinTM稀释液。将GeneTwinTM稀释液加到DNA稀释液中,吹打混匀后,室温放置5 min,获得100 μL转染复合物。最后将转染复合物直接加到细胞培养基中,轻摇细胞板混匀,转染48 h后收样。
1.6 免疫荧光(immunofluorescence)实验
将HeLa细胞接种在玻片上。接种24 h后转染野生型和突变质粒,继续培养48 h后,收细胞进行处理。在室温下用4%多聚甲醛固定液固定30 min,并用0.1% Triton X-100渗透15 min。之后用含5% BSA的PBS封闭30 min,将样品与FLAG抗体(Invitrogen,美国) 4℃孵育过夜,抗体稀释度为1∶500。PBS洗涤3次后,加入荧光二抗(Invitrogen,美国)避光孵育1 h,二抗稀释度为1∶1000,并用DAPI (Invitrogen,美国)染细胞核。最后在正置荧光生物显微镜(ZEISS,德国)下成像拍片。
2017年12月公布的《普通高中语文课程标准》第一次明确了语文学科核心素养的地位与内涵。新课程标准要求语文教育“以核心素养为本,推进语文课程深层次的改革”①,并明确语文核心素养“主要包括语言建构与运用、思维发展与提升、审美鉴赏与创造、文化传承与理解四个方面”②。语文核心素养更为注重学生的主动性,为作文自改教学策略的探索,提供了新的方向。
1.7 蛋白质印记(Western blot)实验
用RIPA裂解缓冲液(强)裂解转染野生型和突变质粒的HeLa细胞,离心后取上清液与4×上样缓冲液混合并在95℃加热使蛋白质变性。通过SDS-PAGE进行蛋白分离,使用湿转法将蛋白质转移到PVDF膜上。然后用5%的脱脂牛奶封闭1 h,将PVDF膜与FLAG抗体4℃孵育过夜,抗体稀释度为1∶1000。最后用TBST (含1% Tween-20)洗涤3次后,用二抗(Invitrogen,美国)室温孵育1 h并进行显影,二抗稀释度为1∶5000。
1.8 细胞增殖(cell proliferation)实验
细胞增殖实验使用细胞计数试剂盒(上海李记科技有限公司,上海)进行检测。使用96孔板进行接种,将转染了野生型质粒或突变质粒的HeLa细胞分别接种到培养板上,每种质粒的细胞接种30个孔,每个孔接种1000个细胞。6 h后,在每种质粒的5个重复孔中分别加入10微升CCK8(cell counting kit)待测药物孵育2 h,用多功能微孔板检测器(BIOTEK,美国)测量每个孔在450 nm处的吸光度。在第一次测量吸光度后的24 h、48 h、72 h、96 h、120 h各检测一次,每次测量5个重复孔。
1.9 统计分析
使用GraphPad Prism 9软件以及统计分析方法Student’s-test和方差分析处理数据,<0.05被认为具有统计学意义。
2 结果与分析
2.1 患者的临床表现
本研究纳入的研究对象主要病例信息如下:
患者1,28岁,女,原发不孕8年。在ICSI (intracytoplasmic sperm injection)中使用降调节短效长方案获得17枚卵母细胞,其中3枚卵退化,剩余14枚卵均不成熟,均停留在MI期,且经体外成熟培养(IVM)后仍未成熟。
患者2,30岁,女,原发不孕4年,尝试了3次体外助孕。第一次体外受精(fertilization,IVF)中使用降调节短效长方案获得2枚卵母细胞,均不成熟;第二次IVF中使用降调节短效长方案获得11枚卵母细胞,其中10枚卵子都未成熟;第三次的ICSI中使用拮抗剂方案获得7枚卵母细胞,均未成熟,其中4枚阻滞在GV期,3枚阻滞在MI期。
患者3,29岁,女,原发不孕2年。在两个IVF周期中,第一次使用拮抗剂方案获得了21枚卵母细胞,其中19枚卵母细胞未成熟,另外2枚卵成熟但未受精;第二次使用拮抗剂方案获得了23枚卵母细胞,均未成熟(表1)。

表1 患者的临床特征
2.2 全外显子组测序结果分析发现患者携带TRIP13基因突变
患者全外显子组测序分析和Sanger验证,发现:家族1的先证者基因9号外显子上携带纯合突变c.859A>G(p.Ile287Val),患者的父母均携带c.859A>G杂合突变,符合常染色体隐性遗传致病模式(图1,A和B)。c.859A>G突变表现为基因9号外显子的859位点的腺嘌呤突变为鸟嘌呤,从而引起错义突变,对应编码的287位的氨基酸由异亮氨酸突变为缬氨酸(p.Ile287Val)。该突变位点位于基因序列的ATP酶结构域上,且该位点氨基酸在6个不同物种中高度保守(图1E);查阅gnomAD数据库,该位点在人群中突变的频率为0(表2);根据ACMG对基因变异的致病性分级标准判定该变异为临床意义未明变异(PM1+PM2+PP3);使用REVEL功能学预测软件进行致病性预测,其结果为0.687,低于预测软件的参考阈值0.75(表3)。家族2的先证者基因1号外显子上携带纯合突变c.77A>G(p.His26Arg),患者父母和兄弟均未携带该位点的突变。c.77A>G突变表现为基因1号外显子的77位点的腺嘌呤突变为鸟嘌呤,从而引起错义突变,对应编码的26位的氨基酸由组氨酸突变为精氨酸(p.His26Arg),该位点的突变已有文献报道[6,16]。该突变位点位于基因序列的N端,且该位点氨基酸在5个不同物种中保守,在小鼠()中不保守;该位点在人群中突变的频率为0;ACMG评级为致病(PM2+PP5+BP1+ BP4);REVEL软件致病性预测结果为0.17,低于预测软件的参考阈值0.75。家族3的先证者基因4和12号外显子上携带复合杂合突变c.409G> A (p.Asp137Asn)和c.1150A>G(p.Ser384Gly)。其中,c.409G>A突变遗传自父亲,c.1150A>G突变遗传自母亲,且父母均为杂合突变,符合常染色体隐性遗传致病模式。c.409G>A突变表现为基因4号外显子的409位点的鸟嘌呤突变为腺嘌呤,从而引起错义突变,对应编码的137位的氨基酸由天冬氨酸突变为天冬酰胺(p.Asp137Asn);c.1150A>G突变表现为基因12号外显子的1150位点的腺嘌呤突变为鸟嘌呤,从而引起错义突变,对应编码的384位的氨基酸由丝氨酸突变为甘氨酸(p.Ser384Gly)。c.409G>A突变位点位于基因序列的N端,且该位点氨基酸在5个不同物种中保守,在鸡()中不保守;该位点在人群中突变的频率为0;ACMG评级为临床意义未明变异(PM2+PP3);REVEL软件致病性预测结果为0.616,低于预测软件的参考阈值0.75。c.1150A>G突变位点位于基因序列的C端,且该位点氨基酸在6个不同物种中高度保守;该位点在人群中突变的频率为0;ACMG评级为临床意义未明变异(PM2+PP3);REVEL软件致病性预测结果为0.823,高于预测软件的参考阈值0.75。通过PolyPhen2和Mutation Taste工具对四个突变位点的致病性预测结果见表3。
PyMol软件对比结果显示这四种错义突变使得TRIP13蛋白结构中氨基酸间的氢键发生了改变;另外,突变p.His26Arg中由于氢键的改变使得该位点氨基酸与与原有相连接的氨基酸断开并且与其他位点的氨基酸形成新的连接,突变p.Asp137Asn和p.Ile287Val中由于氢键的增加使得该位点氨基酸与其他位点的氨基酸形成新的连接。编码TRIP13蛋白的26位氨基酸由组氨酸突变为精氨酸(p.His26Arg),突变前的组氨酸与编码TRIP13蛋白71位氨基酸的谷氨酰胺之间有一个氢键,与72位的丝氨酸有两个氢键,与133位的组氨酸有一个氢键,突变后形成的精氨酸与TRIP13蛋白72位氨基酸的丝氨酸之间的一个氢键断开,与133位的组氨酸相连接的氢键断开,另外与编码TRIP13蛋白71位氨基酸的谷氨酰胺、72位的丝氨酸以及104位的谷氨酸形成新的氢键(图1C);编码TRIP13蛋白的137位氨基酸由天冬氨酸突变为天冬酰胺(p.Asp137Asn),突变前的天冬氨酸与周围氨基酸均未形成氢键,突变后形成的天冬酰胺与编码TRIP13蛋白134位氨基酸的甘氨酸形成新的氢键;编码TRIP13蛋白的287位氨基酸由异亮氨酸突变为缬氨酸(p.Ile287Val),突变前的异亮氨酸只与编码TRIP13蛋白283位氨基酸的谷氨酰胺之间有一个氢键,突变后形成的缬氨酸与编码TRIP13蛋白290位氨基酸的组氨酸形成新的氢键;编码TRIP13蛋白的384位氨基酸由丝氨酸突变为甘氨酸(p.Ser384Gly),突变前的丝氨酸与编码TRIP13蛋白387位氨基酸的缬氨酸之间有两个氢键,与388位亮氨酸有一个氢键,突变后形成的甘氨酸与编码TRIP13蛋白387位氨基酸的缬氨酸之间的一个氢键断开。同时,对比结果显示四个突变对TRIP13蛋白的三级结构都有一定的影响(图1D)。我们推测,这些突变可能通过影响蛋白内部的氢键结合造成了不同程度蛋白结构的改变。
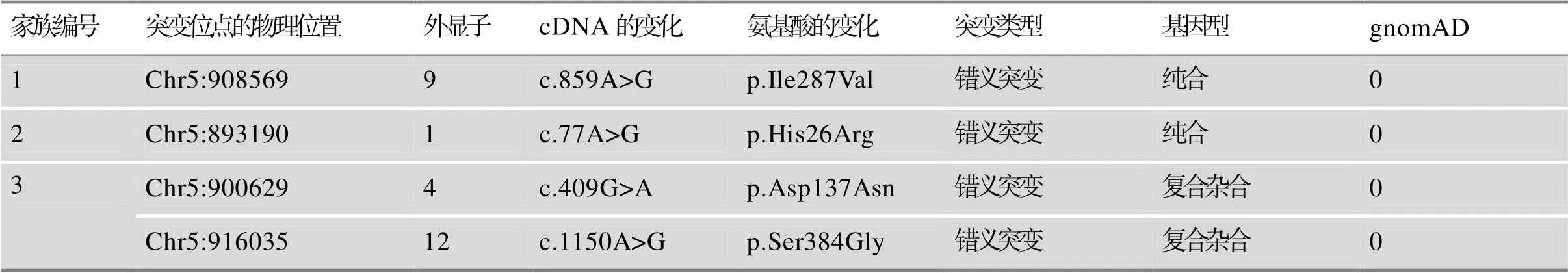
表2 3例患者TRIP13基因突变位点的信息

表3 3例患者TRIP13基因突变位点致病性预测
*变异评级(ACMG分级)是根据美国医学遗传学与基因组学学会(ACMG)发布的最新版基因变异解读标准和指南判定变异评级。
2.3 蛋白定量实验证明突变HeLa细胞TRIP13蛋白表达上升
为了进一步明确基因突变位点的致病性,本研究在HeLa细胞中分别转染了携带FLAG标签的c.77A>G、c.409G>A、c.859A>G、c.1150A>G突变质粒。免疫荧光结果发现这四种突变对TRIP13蛋白在HeLa细胞中的定位未造成明显改变:携带野生型与突变质粒的HeLa细胞中TRIP13蛋白均定位于细胞质(图2A)。此外,免疫印记结果显示转染突变质粒的细胞内TRIP13蛋白丰度都有所上升(图2B)。经进一步定量分析,结果显示转染c.77A>G突变质粒的HeLa细胞中TRIP13蛋白约是转染野生型质粒HeLa细胞中TRIP13蛋白的1.5倍(=0.027);转染c.409G>A突变质粒的HeLa细胞中TRIP13蛋白约是转染野生型质粒HeLa细胞中TRIP13蛋白的1.5倍(=0.0045);转染c.859A>G突变质粒的HeLa细胞中TRIP13蛋白显著增加,约是转染野生型质粒HeLa细胞中TRIP13蛋白的2.5倍(=0.0107);转染c.1150A>G突变质粒的HeLa细胞中TRIP13蛋白显著增加,约是转染野生型质粒HeLa细胞中TRIP13蛋白的2倍(=0.0444)(图2C)。
A:先证者家系图谱;B:家系突变位点基因检测结果;C:TRIP13蛋白氨基酸和氢键的变化,红色名称表示发生突变的氨基酸,黑色箭头表示变化后的氢键,黑色名称表示突变后与该位点相连的氨基酸;D:TRIP13蛋白三级结构预测,黑色箭头指向的位置表示突变氨基酸的位置;E:突变位点保守性分析及基因突变位置。
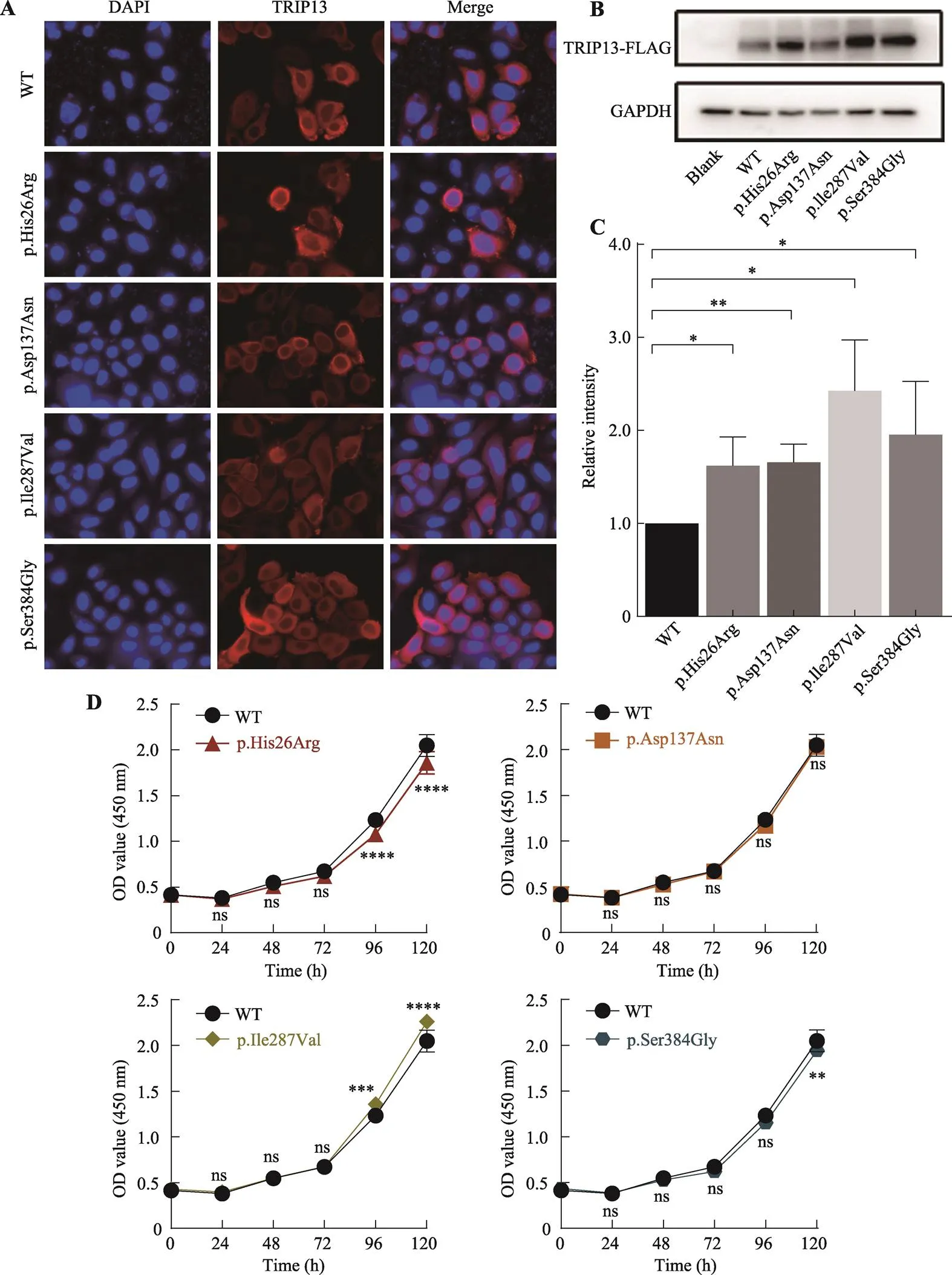
图2 TRIP13基因突变位点致病性分析
A:体外转染不同突变质粒对TRIP13蛋白在HeLa细胞中的定位影响;B:免疫印迹实验检测不同突变对转染的HeLa细胞中TRIP13蛋白水平的影响;C: 图B中TRIP13蛋白表达丰度的相对定量分析。D:细胞增殖实验检测不同突变对HeLa细胞增殖的影响。*:<0.05;**:<0.01;***:<0.001;****:<0.0001。
2.4 细胞增殖实验提示突变HeLa细胞增殖速率异常
有研究报道基因在各种人类肿瘤中高表达,并促进肿瘤细胞的增殖[11,12,17]。为了验证新发现的四个基因突变是否会对细胞增殖产生影响,本研究对转染野生型和突变质粒的HeLa细胞进行细胞增殖实验,结果显示转染c.77A>G突变质粒的HeLa细胞在转染120 h后增殖速度显著下降(<0.0001);转染c.409G>A突变质粒的HeLa细胞增殖速率几乎无变化;转染c.859A>G突变质粒的HeLa细胞在转染120 h后增殖速度显著增加(<0.0001);转染c.1150A>G突变质粒的HeLa细胞在转染120 h后增殖速度有一定的下降(=0.0047)。经进一步将四个突变与对照组在不同时间点对HeLa细胞增殖影响进行两两比较,结果显示转染c.77A>G突变质粒的HeLa细胞在转染72~120 h内与转染野生型质粒相比增殖速度显著下降(<0.0001);转染c.859A>G突变质粒的HeLa细胞在转染72~96 h内细胞增殖速度显著增加(=0.0003),转染96~120 h内细胞增殖速度也显著增加(<0.0001);转染c.1150A>G突变质粒的HeLa细胞在转染96~120 h内增殖速度有所下降(=0.0038)(图2D)。这些结果提示基因突变在一定程度上影响了细胞周期进程。
2.5 TRIP13基因不同位点突变引起不同疾病
结合本文新发现的病例信息,本研究总结了2007~2020年已报道的基因引起不同疾病的突变位点,汇总突变位点的具体信息如表4所示。TRIP13蛋白由432个氨基酸组成,包含N末端,C末端和一个ATP酶结构域。目前总共有13个突变位点,其中4个突变位于N末端,5个突变位于ATP酶结构域以及4个突变位于C末端。涉及到的病种包括:肾母细胞瘤(Wilms tumors);由卵母细胞成熟阻滞导致的女性不孕症;由合子卵裂失败(zygotic cleavage failure,ZCF)导致的女性不孕症。
3 讨论
卵母细胞成熟阻滞是女性原发性不孕的病因之一,在辅助生殖过程中,这类患者的卵母细胞在卵泡刺激素和人绒毛膜促性腺激素给药后仍处于未成熟状态,停留在GV和MI期[19]。已有许多研究报道了与卵母细胞成熟阻滞有关的基因:基因突变会破坏卵母细胞减数分裂微管形成和纺锤体组装,使卵母细胞停滞在MI期,导致女性不孕[20];基因突变会使卵母细胞出现大极体,对称分裂和异常纺锤体等形态学缺陷而使其阻滞在GV期,从而导致女性不孕[7,21,22]。2020年Zhang等[6]报道了五例因基因突变导致人类卵母细胞成熟阻滞的纯合或复合杂合错义致病突变。随后,本课题组在2022年报道了三例因纯合错义突变导致合子卵裂失败的病例[16]。本文发现了来自三例患者基因中的导致人类卵母细胞成熟阻滞的四个突变位点,其中3个突变位点在国内外尚未有文献报道。

表4 TRIP13基因突变位点总结
基因在有丝分裂和减数分裂发生过程中发挥重要作用。在有丝分裂中,TRIP13蛋白可以联合p31comet以及ATP,解聚有丝分裂检查点复合体(mitotic checkpoint complex,MCC),沉默检查点功能,促进有丝分裂进程[23]。TRIP13蛋白在减数分裂中促进带有HORMA结构域的蛋白HORMADs在染色体上的积累和去除,调节DNA断裂的修复和染色体重组过程[24,25]。因此TRIP13可能通过不同途径在有丝分裂和减数分裂中发挥不同作用。此外,与基因有关的人类疾病的报道中,有研究发现基因纯合无义突变或剪接突变会引发肾母细胞瘤[18],且肾母细胞瘤相关的突变导致TRIP13蛋白完全不表达。本研究中发现的导致卵母细胞成熟阻滞的不同位点的错义突变均使得TRIP13蛋白表达增加,而这三名患者并未发现与肾母细胞瘤相关的疾病。这提示不同疾病的产生可能与突变导致的蛋白表达的差异程度有关。另外,基因突变导致女性不孕的表型中,除了卵母细胞成熟阻滞外,也发现了不同的表型:例如基因复合杂合错义突变c.77A>G(p.His26Arg)和c.1258A>G(p. Lys420Glu)导致合子卵裂失败。以上这些研究在一定程度上验证了基因不同位置的突变对蛋白表达和功能的影响会造成个体表型的差异。
是一种AAA+ATP酶,在包括睾丸,卵巢在内的各种组织中表达[25,26]。基因的突变在基因序列的C端和N端以及ATP酶结构域上均有发生,在女性中导致卵母细胞成熟阻滞或者合子卵裂失败。迄今为止已发现5种与卵母细胞成熟阻滞有关的基因突变,分别是c.77A>G(p.His26Arg)、c.518G>A(p.Arg173Gln)、c.907G>A(p.Glu303Lys)、c.592A>G(p.Ile198Val)、c.739G>A(p.Val247Met),以及3种与合子卵裂失败有关的基因突变,分别是c.77A> G(p.His26Arg)、c.1141G>A(p. Glu381Lys)和c.1258A> G(p. Lys420Glu)[6,16]。本文中发现了患者1存在纯合错义突变c.859A>G(p.Ile287Val)以及患者3存在复合杂合突变c.409G>A(p.Asp137Asn)以及c.1150A>G(p.Ser384Gly),均为基因新发现的导致卵母细胞成熟阻滞的致病位点。本研究中在家族2的先证者中发现基因纯合错义突变c.77A>G(p.His26Arg),既往有关的两篇研究中也都曾报道过基因在这一位点的错义突变。这提示基因的第77位碱基可能是基因中导致卵母细胞成熟阻滞的高发位点。总的来说,目前为止发现的所有基因突变造成的不孕表型中,70%的基因突变与卵母细胞成熟阻滞相关,30%的突变导致合子卵裂失败。
基因在各种人类肿瘤中高表达,并在肿瘤的发生和转移中发挥重要功能[11,12]。已有研究证实在膀胱癌组织样本中的基因表达显著升高;体外实验发现基因的过表达会诱导G2/ M期的细胞周期停滞,促进膀胱癌细胞的生长和活力以及集落形成能力,敲低会抑制细胞生长,诱导膀胱癌细胞凋亡[17]。另外也有研究表明与正常卵巢细胞系相比,上皮性卵巢癌细胞系(SKOV-3,HEY和OVCAR-3)中的表达显著上调;体外实验发现沉默上皮性卵巢癌细胞中的可抑制细胞增殖,减少细胞侵袭和迁移[27]。在对细胞增殖的探讨中发现,与转染野生型质粒相比,转染突变质粒的HeLa细胞增殖过程出现不同程度的异常,这说明基因在不同位点的突变确实对细胞周期进程的影响不一致,因此导致的细胞增殖过快或过慢都会影响细胞正常分裂,这可能是造成卵母细胞减数分裂进程阻滞的原因。
卵母细胞成熟阻滞是女性不孕的主要原因之一,目前针对卵母细胞成熟阻滞导致的不孕问题,除了体外成熟培养卵母细胞和接受卵母细胞捐献之外,没有其他有效的治疗手段[28]。越来越多的研究开始关注对突变患者的卵子进行非基因编辑的分子干预,期望能够逆转卵母细胞成熟阻滞的表型。之前的研究报道了将正常的的cRNA注射到基因纯合错义突变患者的卵母细胞中,可以在一定程度上挽救卵母细胞停滞的表型:实验发现所有注射cRNA的MI卵母细胞都排出了第一极体,相比之下,未注射的卵母细胞即使在长期培养后仍处于MI阶段[6]。这些实验结果为携带致病突变的不孕患者提供了一种可能的治疗方法,但未来仍应在小鼠或非人灵长类动物模型中仔细评估其有效性和安全性。
综上所述,本文报道了三例携带基因突变的女性不孕患者,三名患者均表现为卵子不熟,其卵母细胞阻滞在GV或MI期。其中,突变c.859A>G(p.Ile287Val),c.409G>A(p.Asp137Asn)和c.1150A>G(p.Ser384Gly)均为新发现的突变位点。本研究进一步证实了这些突变能够影响TRIP13蛋白表达,使得TRIP13蛋白不同程度的异常增加,进而导致细胞周期进程紊乱。本研究扩充了关于人类卵母细胞成熟阻滞的致病图谱,为女性不孕患者的诊断、遗传咨询提供了重要的依据,同时也将有助于提高人们对人类卵母细胞成熟阻滞致病机制的理解。
[1] Guerri G, Maniscalchi T, Barati S, Gerli S, Di Renzo GC, Della Morte C, Marceddu G, Casadei A, Laganà AS, Sturla D, Ghezzi F, Garzon S, Unfer V, Bertelli M. Non-syndromic monogenic female infertility., 2019, 90(10-S): 68–74.
[2] De Vos M, Grynberg M, Ho TM, Yuan Y, Albertini DF, Gilchrist RB. Perspectives on the development and future of oocyte IVM in clinical practice., 2021, 38(6): 1265–1280.
[3] Fei CF, Zhou LQ. Gene mutations impede oocyte maturation, fertilization, and early embryonic development., 2022, 44(10): e2200007.
[4] Jiao SY, Yang YH, Chen SR. Molecular genetics of infertility: loss-of-function mutations in humans and corresponding knockout/mutated mice., 2021, 27(1): 154–189.
[5] Sang Q, Zhou Z, Mu J, Wang L. Genetic factors as potential molecular markers of human oocyte and embryo quality., 2021, 38(5): 993–1002.
[6] Zhang ZH, Li B, Fu J, Li R, Diao FY, Li CH, Chen BB, Du J, Zhou Z, Mu J, Yan Z, Wu L, Liu S, Wang WJ, Zhao L, Dong J, He L, Liang XZ, Kuang YP, Sun XX, Sang Q, Wang L. Bi-allelic missense pathogenic variants in TRIP13 cause female infertility characterized by oocyte maturation arrest., 2020, 107(1): 15–23.
[7] Cao QQ, Zhao C, Wang CJ, Cai LB, Xia M, Zhang XL, Han J, Xu YY, Zhang JQ, Ling XF, Ma X, Huo R. The recurrent mutation in PATL2 inhibits its degradation thus causing female infertility characterized by oocyte maturation defect through regulation of the mos-mapk pathway., 2021, 9: 628649.
[8] Christou-Kent M, Kherraf ZE, Amiri-Yekta A, Le Blévec E, Karaouzène T, Conne B, Escoffier J, Assou S, Guttin A, Lambert E, Martinez G, Boguenet M, Fourati Ben Mustapha S, Cedrin Durnerin I, Halouani L, Marrakchi O, Makni M, Latrous H, Kharouf M, Coutton C, Thierry-Mieg N, Nef S, Bottari SP, Zouari R, Issartel JP, Ray PF, Arnoult C. PATL2 is a key actor of oocyte maturation whose invalidation causes infertility in women and mice., 2018, 10(5): e8515.
[9] Feng RZ, Yan Z, Li B, Yu M, Sang Q, Tian GL, Xu Y, Chen BB, Qu RG, Sun ZG, Sun XX, Jin L, He L, Kuang YP, Cowan NJ, Wang L. Mutations in TUBB8 cause a multiplicity of phenotypes in human oocytes and early embryos., 2016, 53(10): 662–71.
[10] Cao TQ, Guo J, Xu Y, Lin XF, Deng WF, Cheng LZ, Zhao H, Jiang S, Gao M, Huang JJ, Xu YW. Two mutations in TUBB8 cause developmental arrest in human oocytes and early embryos., 2021, 43(5): 891–898.
[11] Agarwal S, Behring M, Kim HG, Chandrashekar DS, Chakravarthi BVSK, Gupta N, Bajpai P, Elkholy A, Al Diffalha S, Datta PK, Heslin MJ, Varambally S, Manne U. TRIP13 promotes metastasis of colorectal cancer regardless of p53 and microsatellite instability status., 2020, 14(12): 3007–3029.
[12] Zhang GH, Zhu QZ, Fu G, Hou JB, Hu XS, Cao JJ, Peng W, Wang XW, Chen F, Cui HJ. TRIP13 promotes the cell proliferation, migration and invasion of glioblastoma through the FBXW7/c-MYC axis., 2019, 121(12): 1069–1078.
[13] Ma HT, Poon RYC. TRIP13 functions in the establishment of the spindle assembly checkpoint by replenishing O-MAD2., 2018, 22(6): 1439–1450.
[14] Ma HT, Poon RYC. TRIP13 regulates both the activation and inactivation of the spindle-assembly checkpoint., 2016, 14(5): 1086–1099.
[15] Vader G. Pch2(TRIP13): controlling cell division through regulation of HORMA domains., 2015, 124(3): 333–339.
[16] Hu HL, Zhang SP, Guo J, Meng F, Chen XQ, Gong F, Lu GX, Zheng W, Lin G. Identification of novel variants of thyroid hormone receptor interaction protein 13 that cause female infertility characterized by zygotic cleavage failure., 2022, 13: 899149.
[17] Lu SC, Guo MJ, Fan ZM, Chen Y, Shi XQ, Gu CY, Yang Y. Elevated TRIP13 drives cell proliferation and drug resistance in bladder cancer., 2019, 11(7): 4397–4410.
[18] Yost S, de Wolf B, Hanks S, Zachariou A, Marcozzi C, Clarke M, de Voer R, Etemad B, Uijttewaal E, Ramsay E, Wylie H, Elliott A, Picton S, Smith A, Smithson S, Seal S, Ruark E, Houge G, Pines J, Kops GJPL, Rahman N. Biallelic TRIP13 mutations predispose to Wilms tumor and chromosome missegregation., 2017, 49(7): 1148–1151.
[19] Biswas L, Tyc K, El Yakoubi W, Morgan K, Xing JC, Schindler K. Meiosis interrupted: the genetics of female infertility via meiotic failure., 2021, 161(2): R13–R35.
[20] Feng RZ, Sang Q, Kuang YP, Sun XX, Yan Z, Zhang SZ, Shi JZ, Tian GL, Luchniak A, Fukuda Y, Li B, Yu M, Chen JL, Xu Y, Guo L, Qu RG, Wang XQ, Sun ZG, Liu M, Shi HJ, Wang HY, Feng Y, Shao RJ, Chai RJ, Li QL, Xing QH, Zhang R, Nogales E, Jin L, He L, Gupta ML Jr, Cowan NJ, Wang L. Mutations in TUBB8 and human oocyte meiotic arrest., 2016, 374(3): 223–232.
[21] Masciarelli S, Horner K, Liu C, Park SH, Hinckley M, Hockman S, Nedachi T, Jin C, Conti M, Manganiello V. Cyclic nucleotide phosphodiesterase 3A-deficient mice as a model of female infertility., 2004, 114(2): 196–205.
[22] Oh JS, Han SJ, Conti M. Wee1B, Myt1, and Cdc25 function in distinct compartments of the mouse oocyte to control meiotic resumption., 2010, 188(2): 199–207.
[23] Eytan E, Wang KX, Miniowitz-Shemtov S, Sitry-Shevah D, Kaisari S, Yen TJ, Liu ST, Hershko A. Disassembly of mitotic checkpoint complexes by the joint action of the AAA-ATPase TRIP13 and p31(comet)., 2014, 111(33): 12019–12024.
[24] Gu YJ, Desai A, Corbett KD. Evolutionary dynamics and molecular mechanisms of HORMA domain protein signaling., 2022, 91: 541–569.
[25] Roig I, Dowdle JA, Toth A, de Rooij DG, Jasin M, Keeney S. Mouse TRIP13/PCH2 is required for recombination and normal higher-order chromosome structure during meiosis., 2010, 6(8): e1001062.
[26] Li XCL, Schimenti JC. Mouse pachytene checkpoint 2 (trip13) is required for completing meiotic recombination but not synapsis., 2007, 3(8): e130.
[27] Zhou XY, Shu XM. TRIP13 promotes proliferation and invasion of epithelial ovarian cancer cells through Notch signaling pathway., 2019, 23(2): 522–529.
[28] Hatırnaz Ş, Hatırnaz ES, Ellibeş Kaya A, Hatırnaz K, Soyer Çalışkan C, Sezer Ö, Dokuzeylül Güngor N, Demirel C, Baltacı V, Tan S, Dahan M. Oocyte maturation abnormalities - a systematic review of the evidence and mechanisms in a rare but difficult to manage fertility pheneomina., 2022, 19(1): 60–80.
Novel mutations inlead to female infertility with oocyte maturation arrest
Xiangjiang Lv1, Jing Guo2, Ge Lin1,2
Oocyte maturation arrest (OMA) refers to a rare clinical phenomenon of oocyte maturation disorder caused by abnormal meiosis, which is also one of the primary causes of female infertility. The clinical manifestations of these patients are often characterized with failure to obtain mature oocytes after repeated ovulation stimulation and/or inducedmaturation. To date, mutations in,andhave been demonstrated to be associated with OMA, but studies on the genetic-based factors and mechanisms of OMA are still incomplete. In this study, peripheral blood from 35 primary infertile women characterized with recurrent OMA during assisted reproductive technology (ART) were subjected to whole-exome sequencing (WES). By using Sanger sequencing and co-segregated analysis, we identified four pathogenic variants in. Proband 1 had a homozygous missense mutation of c.859A>G appeared on the 9th exon, which resulted in substitution of Ile287 to valine (p.Ile287Val); proband 2 had a homozygous missense mutation of c.77A>G on the 1st exon, which resulted in substitution of His26 to arginine (p.His26Arg); and proband 3 had compound heterozygous mutations of c.409G>A and c.1150A>G on the 4th and 12th exon, which resulted in the substitutions of Asp137 to asparagine (p.Asp137Asn) and Ser384 to glycine (p.Ser384Gly) in the encoded protein respectively. Three of these mutations have not been reported previously. Further, transfection of plasmids harboring the respective mutatedin HeLa cells resulted in changes in TRIP13 expression and abnormal cell proliferation as demonstrated by western blotting and cell proliferation assay respectively. This study further summarizes themutations reported previously and expands the mutation spectrum ofpathogenic variants, thereby providing a valuable reference for further research on the pathogenic mechanism of OMA associated withmutations.
oocyte maturation arrest;; meiosis; female infertility
2023-02-01;
2023-03-30;
2023-04-21
国家自然科学基金项目(编号:81901473)和中信湘雅生殖遗传专科医院院内项目(编号:YNXM-201810)资助[Supported by the National Natural Science Foundation of China (No. 81901473), and the Scientific Research Foundation of Reproductive and Genetic Hospital of China International Trust Investment Corporation (CITIC) Xiangya (No. YNXM-201810)]
吕香江,在读硕士研究生,专业方向:遗传学。E-mail:lvxiangjiang@163.com
林戈,博士,研究员,研究方向:遗传学。E-mail: linggf@hotmail.com
郭静,博士,副研究员,研究方向:生殖医学。E-mail: 815457238@qq.com
10.16288/j.yczz.23-022
(责任编委: 谷峰)
