非劣效性设计临床试验
2023-07-03周齐严鸿伊张艳丽潘岳松
周齐,严鸿伊,张艳丽,潘岳松
当临床需要评估新药、改良术式或新干预措施的疗效时,通常需将这些新治疗方法与原有的某种具有公认疗效的经典治疗方法做比较,通常情况下,使用优效性设计试验检验新的治疗方法是否优于原有治疗方法。但是,随着医学水平的进步,利用优效性检验获得更佳疗效药物的难度越来越大,这就促使研究者将注意力开始转向标准治疗手段以外方面的改善[1]。非劣效性检验的主要研究目的是验证新治疗方法的疗效不比对照组的疗效差,或者疗效略差但是并没有超过临床上有意义的程度,适用于具有客观疗效指标的临床研究。目前非劣效性设计临床试验已经成为评估药物、器械、生物制剂和其他干预措施的主要工具[2]。
本文简要介绍非劣效性设计临床试验的基本概念、历史渊源、设计要点及其与等效性和优效性设计的比较,并进行具体案例解读,以帮助相关领域研究者正确认识、实施和评价非劣效性设计方法。
1 非劣效性设计临床试验的概念和历史渊源
从1948年世界上第1项设计规范的随机、双盲对照临床试验——链霉素治疗结核试验发布后,对新药物的探索逐渐成为了临床研究的重点和热点。传统的随机对照试验(randomized clinical trials,RCT)多是以安慰剂作为对照组的优效性检验,在临床中被视为药物开发的金标准。然而,由于很多药物疗效不断被证实,此类优效性研究的发展逐渐受限于伦理质疑或开发更佳疗效新药的技术难度[3]。随着临床研究对新药物或新技术的不断探索,研究者开始关注疗效外的其他层面效果,例如:药物的安全性、不良反应、疗程以及治疗费用等,如果新治疗方法在这些方面相较于原方法表现更加优秀且更易被患者接受,即使新方法较经典治疗方法在疗效上没有表现出明显优势或疗效差异在临床可接受范围内,这都说明新治疗方法仍适合进一步开发。非劣效性设计即是适用于这种情况的新的临床试验方法[4]。20世纪70年代末,以Dunnett和Gent等[5]为代表发布的第1批非劣效性或等效性设计的研究开始逐渐走进研究者的视线,此时这类研究主要目的是检验仿制药物与标准药物的生物等效性。现阶段非劣效性设计临床试验中,研究者既期望能在一定程度上维持疗效,也希望能发现新药物比标准药物在其他方面的改善,例如:更少的不良反应、更好的使用便利性以及更易被接受的价格等。这类方法相比于优效性检验更加符合伦理要求,在多种潜在获益方面也更具价值,这使得此类设计方法逐渐成为药物临床试验的主要方法之一。
2 非劣效性设计临床试验要点
2.1 适用场景 非劣效性设计临床试验主要适用于以下情况:①开发在疗效上超越经典药物的新药的可能性较低;②新研发的药物与标准药物的疗效相当,但在安全性、成本、患者依从性方面可能更具优势;③使用安慰剂对照不符合伦理要求。
2.2 前提假设与研究目的 非劣效性设计临床试验中的阳性对照组是阳性对照药物或标准药物,其中关键的恒定假设是在研究背景相近前提下,当前研究中阳性对照药物或标准药物具有相比于历史安慰剂的预期疗效。非劣效性设计临床试验的目的是检验试验组的治疗效果不比阳性对照组的治疗效果差,或疗效稍差于阳性对照组但差异仍在临床可接受的范围内[方案事先指定的非劣效界值(∆)][6]。
2.3 对照选择 非劣效性设计临床试验中阳性对照组应是当前临床上针对目标疾病普遍使用、有明确药理作用的药物,并且这类药物的治疗效果已被可获得的、设计良好的临床试验所证实。需要注意的是,非劣效性设计临床试验应重点关注阳性药物的适应证、主要疗效指标和剂量以及适应证人群等方面与此类研究和历史安慰剂研究是否具有一致性[7]。
2.4 研究终点 非劣效性设计临床试验的研究终点应包括以下几个特性:①相对客观,避免主观因素造成的偏倚;②易于测量且不易缺失,以保证数据能尽量被完整、准确地收集记录;③能在既往阳性对照药物和安慰剂对照比较的临床研究中找到该研究终点的相应历史数据,为确定科学的非劣效界值提供依据。
对主要研究终点的评估可采用相对度量和绝对度量。相对度量包括率比、风险比、优势比;绝对度量主要是指均值差、率差和风险差等。当两种度量类型的分析结果一致时,更有利于得出非劣效性的结论[6]。另外,结局指标分为高优和低优两种,其中高优指标是指结局指标的数值越大,说明治疗手段疗效越好,如有效率、治愈率等;低优指标是指结局指标的数值越小,说明治疗手段疗效越好,如死亡率、复发率等[8]。
2.5 样本量计算 非劣效性设计临床试验中样本量计算通常依据主要疗效指标,样本量的估计与试验中事件发生率或合并方差相关,通过不同属性的疗效指标计算样本量的公式不同。具体如下[6,8-9]。
率差的非劣效样本量计算公式:
其中T为试验组率的估计值,C为阳性对照组率的估计值,δ=C-T≥0(高优指标),δ=T-C≥0(低优指标),Z为检验统计量,α为Ⅰ类错误概率,β为Ⅱ类错误概率,∆为非劣效界值,K为两组样本量比例,Nc为对照组样本量,K×Nc为试验组样本量。
率比的非劣效样本量计算公式:
公式中的δ=ln(C-T)≥0(高优指标),δ=ln(T-C)≥0(低优指标),T为试验组率的估计值,C为阳性对照组率的估计值,Z为检验统计量,α为Ⅰ类错误概率,β为Ⅱ类错误概率,∆为非劣效界值,K为两组样本量比例,Nc为对照组样本量,K×Nc为试验组样本量。
均数差的非劣效样本量计算公式:
其中σ2为合并方差的估算值,δ=C-T≥0(高优指标),δ=T-C≥0(低优指标),T为试验组均数的估计值,C为阳性对照组均数的估计值,Z为检验统计量,α为Ⅰ类错误概率,β为Ⅱ类错误概率,∆为非劣效界值,K为两组样本量比例,Nc为对照组样本量,K×Nc为试验组样本量。
2.6 非劣效性界值的确定 非劣效界值(∆)是指试验药物与阳性对照药物相比在临床上可接受的最大疗效损失,这个损失的范围应不超过阳性药物相比于安慰剂对照的临床获益,以保证该研究具有足够的检验灵敏度[6,9]。针对许多规则不明确的非劣效性设计临床试验缺少基于阳性对照药物的历史数据而导致∆值多具有临床主观性的现状,人用药品技术要求国际协调理事会(The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)在临床试验中对照组的选择和相关问题中指出,∆值的确定取决于统计推断和临床判断[10],在设计研究方案时应将设计良好的历史阳性药物和安慰剂对照临床试验数据列表,整理各资料中率的点估计值、区间估计与样本量,考察各数据来源及研究背景(时间、地点、适应证等)以及所采用的统计汇总方法,即所谓的疗效一致性[11]。
确定∆值的常用方法有点估计法、固定界值法、综合法以及德尔菲法。其中固定界值法和综合法是根据阳性对照药物与安慰剂、试验组与阳性对照药物、临床上可接受的最大疗效损失比例之间的相互关系来进行的,是目前美国食品和药品管理局较为推荐的方法。∆值必须在试验设计阶段就依据临床意义、安全评价、研究成本和患者接受难易程度等多方面因素来确定并在试验方案中详细阐明制定标准,在揭盲之前允许修改但必须阐述理由,揭盲后则不允许再次修改[12]。
在非劣效性设计临床试验中,∆值不能高于最小临床意义差异(minimal clinically important difference,MCID)。MCID可以从不同角度进行解读。从医师角度分析,是指对临床治疗有指导意义的最小治疗受益;从患者角度分析,是指不同患者在同一时间点的健康状况差别。常用计算方法有效标法、分布法、专家意见法和文献分析法[13]。然而,目前关于MCID还缺少统一且适用于不同情况的判断标准,需要由专业的学术委员会作为桥梁,联合医疗机构和生物统计专家共同制定。
2.7 净获益程度的计算方法 非劣效性设计临床试验开展前,需要先确定既往阳性对照与安慰剂相比的疗效差异,即阳性对照药物疗效的净获益程度(M)。可以由以下几种方法获得M值。①既往非劣效临床研究:需考虑相似药物在既往研究中的适应证、目标人群、主要终点指标、环境条件等与当前研究的可比性,在得到比较肯定的结果以及临床专家评估后,方可沿用既往研究中的∆值;②既往单项随机、安慰剂对照的优效性研究:对照组疗效的变异性需要考虑在内,建议选用研究结果的95%或者更宽的CI下限作为对照组在既往研究中的疗效大小,以此谨慎评估其在当前研究中的疗效;③多项既往阳性对照与安慰剂或无治疗相比的研究:需使用meta分析方法估算出阳性对照药物相对于安慰剂的疗效差异,以及评估不同研究间的变异性。通过恒定假设评估阳性对照药物在当前研究和既往研究中的疗效一致性后,经过再估算的当前研究中对照组疗效的净获益程度M值称为M1[8-9]。而∆值,又称临床上可接受的最大损失疗效(M2),计算该数值时,应同时将M1度量类型和保留阳性对照疗效适当比例(f)考虑在内[14]。当M1为绝对度量时,M2=(1-f)×M1;若M1是相对度量,则M2=exp[(1-f)ln(M1)]。在临床研究中,通常f的取值范围是0.5~0.8,在心血管药物的非劣效研究中,f常取值0.5。通常情况下,当疗效指标为低优指标时,M1为95%CI上限的倒数;当疗效指标为高优指标时,M1为95%CI下限的倒数。例如:在通过meta分析对历史数据进行分析后,阳性对照药物较安慰剂对照的卒中复发风险(低优指标)减少40%,此时安慰剂/阳性对照=1/(1-0.4)=1.67,依据专家咨询结果取M1=1.5<1.67,取f=0.5,则M2=exp[(1-0.5)ln(1.5)]=1.225。
2.8 统计分析与结果解读 非劣效性设计临床试验的有效性检验需要同时分析更接近真实世界的意向性分析集(intention-totreat,ITT)和更接近于理想状态的符合方案集(per-protocol sets,PPS)。若两个分析集的研究结论一致,说明研究质量控制良好,研究结果可靠;若两个分析集得出的结论不一致,则应进一步讨论和分析[15]。
非劣效性设计临床试验中使用的是单侧检验,需要根据研究终点事件是高优还是低优指标选择比较的CI上下限[16]。以主要研究终点(低优指标)的率比为例,非劣效性设计临床试验有多种可能结果(用双侧95%CI表示)[2,6]:①如果试验组与对照组的不良事件发生风险的CI完全<1,则说明该研究结果为非劣效性或者考虑为优效性。②CI范围跨1,但完全没有超过预先设定好的界值∆,则该研究满足非劣效性的结论。③CI完全没有超过预先设定好的界值,但CI上下限均>1,此时有研究者认为可以得出非劣效的结论,因为CI不包含∆值满足非劣效的假设检验。但是从CI下限来看,试验组的疗效较阳性对照组差,考虑结果可能为样本量不足所致,建议扩大样本来证实结论。④当CI包含∆值但同时跨过1时,表明研究效力不足,只能得到不确定的研究结论。⑤当CI包含∆值但整体>1时,不同学者的观点不一致。有学者认为CI不包含1,且结果显示阳性对照组疗效更好,可以得出劣效性结论[2]。但是根据试验报告统一标准(consolidated standards of reporting trials,CONSORT)声明[17],此时因不能确定疗效差异是否小于非劣效界值但又>1,结论尚不能确定。⑥当CI完全在∆值右侧时,表明试验组疗效劣于阳性对照组。图1对不同结果进行了示例和解读说明。
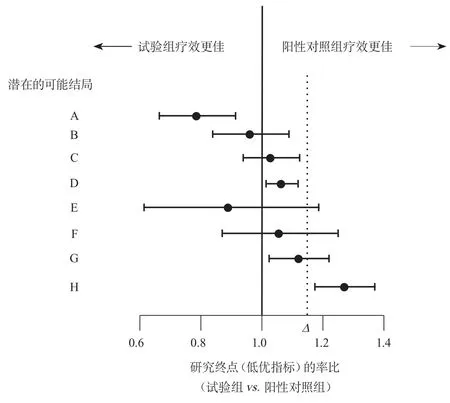
图1 非劣效性设计临床试验可能出现的结果及解读
2.9 报告规范 非劣效性和等效性随机试验的报告规范可参考CONSORT 2010声明的扩展版。该扩展版是在2010年CONSORT声明和2008年CONSORT摘要报告声明的基础上提出的,用于报告非劣效性和等效性试验的CONSORT清单的更新扩展内容,并对与2010年CONSORT清单不同的项目提供了说明性示例和解释,以提高非劣效性和等效性试验的透明度并规范其报告,改善此类试验结果的可解释性以及方法和结论的可重复性,使读者能够评估其结果和结论的可靠程度[17]。
2.10 优势和局限性 在非劣效性设计临床试验中,研究终点可以是多种类型的,如二分类变量、有序变量、连续变量等,这种灵活性有利于非劣效性设计临床试验的广泛应用;另外,非劣效性设计临床试验是以治疗方法和适应证为基础的,在追求试验药物疗效不劣于现有标准药物的同时,注重探索其在疗效之外的优点,比安慰剂对照的临床试验更具有伦理价值。
非劣效性设计临床试验的局限性:非劣效性设计所参考的阳性对照药物历史研究中,对照组并不总是理想的安慰剂对照,致使恒定假设难以验证,检测灵敏度难以保证[9];结果解释的复杂性会增加临床研究各相关方对此类研究方法概念、设计及统计方法的理解难度;∆值过大以及阳性对照药物相对于安慰剂的历史数据存在发表偏倚、统计显著性偏倚及选择偏倚等缺陷,以及生物本身的进化过程都将提高生物爬行现象风险(生物爬行现象是指在连续应用非劣效试验进行药物或器械注册申报时,上一代非劣效试验的试验药物或器械在下一代非劣效试验中被作为阳性对照,经过多代试验后,试验药疗效可能逐渐接近安慰剂的现象)[18]。
3 非劣效性设计与等效性及优效性设计的比较
非劣效性、等效性和优效性设计在适用场景、前提假设与研究目的、对照选择、研究终点、样本量计算、分析人群方面均有不同程度的差异,三者都有各自的优势和局限性。临床研究者需要根据研究目的选择合适的研究设计方法。这3种研究设计的特点比较见表1。

表1 非劣效性设计、等效性设计及优效性设计的比较
4 案例解读
4.1 案例1:TRACE-2试验——一项多中心、开放标签、终点盲法、随机对照、非劣效设计试验 在缺血性脑血管病再灌注的临床治疗中,阿替普酶静脉溶栓是唯一具有确切循证医学证据支持的静脉溶栓药物,但在临床中,阿替普酶溶栓后血管再通率和患者预后效果并不理想;另外,该药必须使用输液泵进行操作,不够便捷。上述问题促使研究者开始寻找替代药物,其中替奈普酶是目前热门的候选药物之一。
由首都医科大学附属北京天坛医院王拥军教授牵头完成的替奈普酶再灌注治疗急性缺血性脑血管病事件Ⅱ(tenecteplase reperfusion therapy in acute ischemic cerebrovascular event Ⅱ,TRACE Ⅱ)研究首次在亚洲急性缺血性脑血管病患者群体中进行了替奈普酶与阿替普酶的对比分析[19]。该研究共纳入来自中国53个研究中心的1430例发病4.5 h内、NIHSS评分5~25分的缺血性卒中患者,按照1∶1的比例随机接受替奈普酶(0.25 mg/kg,最大剂量25 mg)或阿替普酶(0.9 mg/kg,最大剂量90 mg)治疗,研究的主要终点和主要有效性评价指标为90 d mRS评分(0~1分为功能结局良好)。在这项非劣效性设计临床试验中,研究者依据既往同样以良好功能结局为主要终点的阿替普酶与安慰剂药效对比的历史数据,经过专家评估后确定阳性对照药物相比于安慰剂发生结局事件的RR为1.24,95%CI为1.14~1.36,主要研究终点属于高优指标,M1取CI下限的倒数,保留阳性对照疗效比例f取0.5,通过公式计算得到∆值=exp[(1-0.5)×ln(1/1.14)]=0.937。最终在有效性评估中,替奈普酶组705例中的439例(62%)患者和阿替普酶组696例中的405例(58%)患者达到主要终点(RR1.07,95%CI0.98~1.16)。鉴于∆值低于RR的95%CI下限,该研究最终得到替奈普酶非劣效于阿替普酶并且安全性良好的结论。结合替奈普酶具有一次性给药、快速给药的便利优势和良好的患者接受程度,研究结果为急性缺血性卒中患者的溶栓药物选择提供了崭新的“中国思路”。
4.2 案例2:INSURE研究—— 一项随机、双盲、对照、非劣效性试验 在临床实践中,依据指南推荐使用阿司匹林对中重度缺血性卒中患者进行二级卒中预防时,其带来的胃肠道反应和出血是不可忽视的问题,因此各国学者都在寻找其他可替代阿司匹林的二级卒中预防药物。首都医科大学附属北京天坛医院王拥军教授牵头的多中心临床试验——吲哚布芬对比阿司匹林治疗急性缺血性卒中(indobufen versus aspirin in acute ischemic stroke,INSURE)研究是一项随机、双盲、对照、非劣效性试验,目的是探讨吲哚布芬降低中重度缺血性卒中患者90 d新发卒中风险是否非劣效于阿司匹林[20]。这项研究涉及中国163家医院,共纳入5438例发病72 h内的急性中重度缺血性卒中(NIHSS评分4~18分)成年患者。将受试者按照1∶1的比例随机分到吲哚布芬组(100 mg,2次/日)和阿司匹林组(100 mg,1次/日),治疗时间为90 d。主要疗效指标为90 d内新发卒中,主要安全性终点为根据链激酶和t-PA在冠状动脉闭塞全球应用(global utilization of streptokinase and tissue plasminogen activator for occluded coronary arteries,GUSTO)研究定义的90 d内中度或重度出血。该研究预先设定的非劣效性界值为1.25,而主要研究结果使用HR的95%CI的单侧上限是否大于非劣效界值来评估吲哚布芬对比阿司匹林的非劣效性。在最终的分析结果中发现,90 d内吲哚布芬组和阿司匹林组分别有213例(7.9%)和175例(6.4%)患者新发缺血性或出血性卒中(HR1.23,95%CI1.01~1.50),CI上限大于非劣效性界值1.25,非劣效性P=0.44。
应该正确看待INSURE研究未取得非劣效结论的结果。首先,在统计学上非劣效和劣效的界限定义并不统一,未达到非劣效并不意味着是劣效[17],未达到非劣效与劣效不能混为一谈。其次,在INSURE研究中主要终点指标HR的CI下限(1.01)>1,是否可以肯定地认为吲哚布芬劣效于阿司匹林呢?关于这一点,目前是有一定争议的。有学者认为这种情况下可以直接得出劣效的结论,但也有学者认为,根据CONSORT声明中的解释[17],这种情况并不能得到确定性的结论,即不能肯定地认为在中重度卒中二级预防中吲哚布芬劣效于阿司匹林。
非劣效性设计方法是建立在试验药物和阳性药物比较结果假设基础上的一种研究方法,尤其是当临床试验结果在差异性检验中未发现阳性结果时,研究者可以考虑非劣效性设计方法来探索新的治疗手段的其他优点。在考虑应用非劣效性设计临床试验时,科研工作者需要遵守保持此类设计方法有效性的基本原则,进而应用设计良好的非劣效性设计临床试验来探寻具有临床价值的创新性替代方案。
