镫骨强直伴拇指(趾)宽大综合征家系NOG基因新发突变鉴定及临床特征分析
2023-06-08杨长亮黄成成曹婧媛陈睿尧
张 钊, 卢 宇, 杨长亮, 阳 光, 王 丽, 彭 婉,王 超, 黄成成, 曹婧媛, 陈睿尧, 孙 艺
耳聋是造成人类残疾的常见原因,随着分子生物学和遗传学研究的深入,遗传性耳聋逐步被人们重视。根据是否合并其他系统的病变,可分为综合征型耳聋和非综合征型耳聋[1]。镫骨强直伴拇指(趾)宽大综合征(stapes ankylosis with broad thumbs and toes,SABTT)(OMIM#184460)是一种导致综合征型耳聋的常染色体显性遗传疾病,其特征是继发于镫骨强直的双侧传导性听力损失、明显的远视、宽拇指(趾)[2-3]。该综合征的发病与NOG基因突变相关。NOG基因(OMIM#602991)定位于17q22的D17S790~D17S794之间,只有一个外显子(https://www.genenames.org)。Noggin由NOG基因编码,是一种由232个氨基酸残基组成的分泌蛋白[3],是一种骨形态发生蛋白(bone morphogenetic protein,BMP)功能拮抗剂,通过结合和掩盖位于BMP上的Ⅰ型和Ⅱ型受体位点与BMP-7相互作用[4-5]。BMP和Noggin之间的平衡对于骨骼的正常形成至关重要[6-8]。NOG基因突变可能影响Noggin蛋白的分泌、二聚体的形成以及与BMP的结合能力[9]。NOG基因中已确定的突变与表型相关,传导性听力损失、多发的骨发育异常、关节粘连是这种综合征患者的常见症状。本研究分析了被诊断为传导性听力损失的先证者的临床表现,对患者家系的样本进行了生物学分析,致病突变是NOG基因的新杂合突变,诊断为SABTT。报告如下。
1 资料与方法
1.1家系临床资料 家系的先证者是一名34岁女性,湖北武汉人,因“双耳听力下降30余年”在我院耳鼻咽喉头颈外科就诊。对先证者进行了体格检查、听力学测试,包括纯音听阈测定(频率250~8 000 Hz)、声导抗鼓室压图、手和脚的X线、颞骨CT检查。对其父母进行详细的病史询问、体格检查、纯音测听、声导抗检查,并采集外周血样本10 ml。本研究经医院伦理委员会批准(批号:[2018]016-1)。所有参与研究的家系成员签署知情同意书。
1.2研究方法
1.2.1 目标序列捕获与测序 用先证者的静脉血10 ml,标准流程提取基因组DNA(MagPure Buffy Coat DNA Midi KF Kit)。将基因组DNA打断并制备文库,然后通过BGI V4芯片对目标基因外显子及邻近剪切区的DNA进行捕获和富集,最后使用MGISEQ-2000测序平台进行突变检测。测序数据质控指标为:目标区域平均测序深度≥180X,其中目标区深度>20X的位点所占比例>95%。
1.2.2 数据分析 通过BWA将测序的片段与hg19人类参考基因组进行比较,然后删除重复项。GATK用于碱基质量校正SNV、INDEL和基因型检测。通过ExomeDepth检测外显子水平的拷贝数变化。生成目标区域的碱基多态性结果,然后与数据库(NCBI dbSNP、gnomAD)进行比对,对发现的可疑突变进行注释和筛选。
1.2.3 Sanger测序 根据全外显子组测序生物信息学分析结果对NOG基因致病突变进行验证,经先证者及家系成员Sanger测序证实该位点是否在家系中共分离。
1.2.4 致病性分析 根据美国医学遗传学与基因组学会(American Society for Medical Genetics and Genomics,ACMG)指南,对检测出的突变位点进行致病性分析。
2 结果
2.1家系临床特征 先证者女性,34岁,3岁时由父母发现其双耳听力下降,言语发育正常,无眩晕、耳鸣,无耳闷、耳道流脓等症状,双手小指远端指间关节不能屈曲,弱视及远视,生长及智力发育正常。查体:眼裂窄,眼距正常,半圆柱形鼻,鼻翼发育不良(见图1ⓐ),双侧拇指及小指短小,双拇趾宽且短,第1、2脚趾间隙过深,扁平足(见图1ⓑ),双耳廓无畸形,双外耳道通畅,双鼓膜完整,标志清晰。辅助检查:双耳纯音听阈测定提示双耳传导性聋,气骨导差平均50 dB(见图1ⓒ)。声导抗:鼓室压图双耳A型。镫骨肌反射双耳均未引出。颞骨CT显示听骨形态正常,听骨链完整。双手X线正位片:双侧拇指远节短小,双侧小指中节指骨短小畸形,中节与近节指关节融合,双侧第5掌骨短小。双足X线正位片:双侧第1跖骨短小,右侧较明显,扁平足(见图1ⓓ)。该家系2代,共计成员3人,绘制家系图(见图2ⓐ)。仅先证者表现为传导性听力损失,听力损失的特点:幼年发病,随着年龄增长听力逐渐下降,并伴有骨发育异常,弱视及远视。先证者父母均无症状。家系成员临床特征见表1。
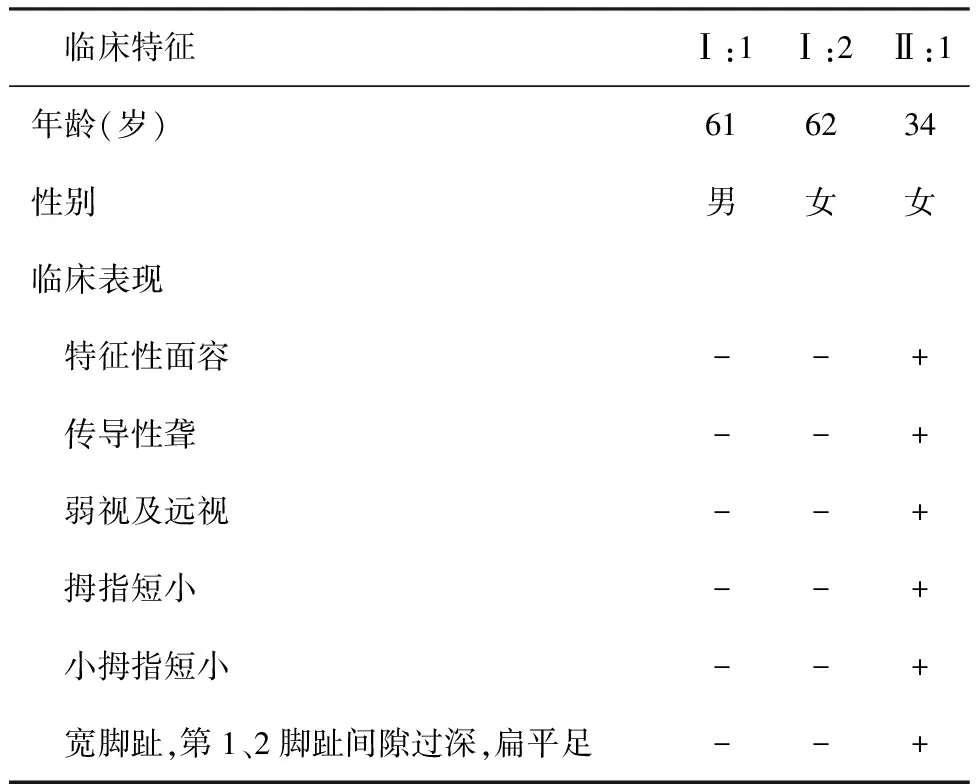
表1 家系成员临床特征

ⓐ面部:眼裂窄,眼距正常,半圆柱形鼻,鼻翼发育不良;ⓑ双手及双脚:双侧拇指及小指短小,双拇趾宽且短,第1、2脚趾间隙过深,扁平足;ⓒ听力:双耳传导性聋,气骨导差平均50 dB;ⓓ双手X线正位片:双侧拇指远节短小,双侧小指中节指骨短小畸形,中节与近节指关节融合,双侧第5掌骨短小。双足X线正位片:双侧第1跖骨短小,右侧较明显,扁平足

ⓐ家系图;ⓑ二代测序结果:先证者(Ⅱ:1)检出NOG基因的一个杂合突变c.679G>T,其父母(Ⅰ:1,Ⅰ:2)该位点均为野生型;ⓒNOG基因的保守性分析显示,该突变位点在多物种之间是高度保守的
2.2NOG基因突变分析结果 二代测序结果显示,先证者(Ⅱ:1)检测出NOG基因的一个杂合突变NM_005450.4:c.679G>T(p.Glu227Ter)。在其他无疾病表型成员中,未发现该位点突变(见图2ⓑ)。对NOG基因编码的Noggin蛋白进行保守性分析,发现其在多物种之间是高度保守的(见图2ⓒ)。多种统计方法预测突变会对基因或基因产物造成有害影响(SIFT,Mutation Taster,Condel,PhyloP Vertebrates,PhyloP Placetal Mammals,GERP++)。
3 讨论
3.1本研究报道了一个以双耳传导性聋为首诊症状的家系,先证者临床表现双侧传导性聋并伴特征性面容、拇指(趾)发育异常、弱视及远视,无眩晕等前庭功能障碍,无耳毒性药物及环境噪声接触史,否认外伤史。通过临床资料分析符合诊断SABTT。
3.2对家系先证者进行高通量测序,并根据《ACMG遗传变异分类标准与指南》进行致病性分析,检测出NOG基因外显子第679位碱基由鸟嘌呤(G)突变为胸腺嘧啶(T),导致其编码的第227位的谷氨酸(Glu)突变为终止密码子(Ter),出现截短突变(PVS1)。该突变是新发现的突变,在gnomAD数据库中未见报道(PM2)。对NOG基因编码的Noggin蛋白进行保守性分析,发现该突变位点在多物种之间高度保守。携带该突变的先证者表型高度符合SABTT的诊断(PP4)。家系验证患者父母没有检测出该突变提示为新生突变(PS2),突变位点是“致病的”。
3.3NOG基因编码Noggin蛋白,是BMP信号传递的关键调控因子,在人体发育特别是软骨和骨骼的发育过程中起重要作用[10-11]。NOG突变根据不同的临床表现诊断为不同的综合征,如SABTT、近端指(趾)关节融合症(proximal symphalangism,SYM1)、多发性骨性连接综合征1型(multiple synostoses syndrome type 1,SYNS1)。几种不同综合征有部分的重叠特征,最常见的包括骨骼和关节形成异常,传导性听力损失,还可能导致半圆柱形鼻等特征性容貌。但SABTT与SYM1和SYNS1的不同之处在于大部分患者都有远视及弱视,但缺乏近端指(趾)关节融合、腕骨和跗骨的融合[12-15]。因此,Potti等[16]提出了一个新的术语:NOG相关系谱障碍(NOG-related-symphalangism spectrum disorder,NOG-SSD),以帮助临床识别和评估所有具有这些表型的患者。这些NOG基因突变可在数据库中获得(https://NOG.lovd.nl)。
3.4目前,文献报道了9个NOG基因突变导致SABTT的家系。Hilhorst-Hofstee[12]及Weekamp[17]分别报道了2个错义突变,即c.103C>T(p.Pro35Ser)和c.608T>C(p.Leu203Pro);Weekamp[17]、Brown[14]和Thomeer[18]分别报道了4个移码突变,即c.129_130dup(p.Val44Glyfs*19)、c.561del(p.Glu188Argfs*76)、c.252dup(p.Glu85Argfs*97)和c.304del(p.Ala102Argfs*22);Brown[14]、Thomeer[18]和Nakashima[19]分别报道了3个无义突变,即c.328C>T(p.Gln110Ter)、c.391C>T(p.Gln131Ter)和c.645C>A(p.C215Ter)。值得注意的是,既往研究报道的与SYM1、SYNS1相关的NOG突变通常是错义突变,而目前已知与SABTT相关的NOG基因的9个突变中,有7个是失活突变,表明NOG基因单倍体表达不足可能特异性地导致SABTT表型出现。Noggin蛋白的C端区域有6个保守半胱氨酸,通过3个二硫键形成环状胱氨酸结构(CCKⅠ-Ⅵ),包括Ⅰ(p.Cys155)-Ⅳ(p.Cys192)、Ⅱ(p.Cys178)-Ⅴ(p.Cys228)和Ⅲ(p.Cys184)-Ⅵ(p.Cys230),形成同源二聚体[20]。本研究中新发现的NOG基因突变,也是一个无义突变,但因靠近编码区末端,可能不会引起无义突变介导的mRNA降解。该突变可能导致Noggin蛋白末端提前5个氨基酸残基发生终止,Noggin蛋白末端区域是高度保守非重复序列,且p.Cys228和p.Cys178可以形成二硫键。推测p.Glu227Ter突变阻止了二硫键的形成,导致功能缺陷。
综上所述,本研究收集到一个诊断为SABTT的家系并明确了家系的致病基因,即NOG基因,鉴定NOG基因一个新的突变位点c.679G>T(p.Glu227Ter)。Nakashima等[19]从病理研究中证实,SABTT患者出生后镫骨融合会逐渐恶化,导致传导性听力损失进行性加重。SABTT患者主要的诉求是改善听力,通常到耳鼻喉科首诊,临床医师需要与耳硬化症等其他传导性聋疾病鉴别[21]。本研究通过临床诊断和分子诊断相结合的方法提高了对此罕见病的认识,为此病的鉴别诊断和家系的遗传咨询提供了依据。
