HPLC-MS/MS 同位素内标法测定水产品中6 种喹诺酮药物残留量
2023-04-18常波
常 波
(营口市农业农村综合发展服务中心(营口市农产品质量安全检验监测中心),辽宁营口 115004)
水产养殖是一个不断增长的全球性行业,随着产量提高而不断增加的兽药使用量已成为全球关注的问题。其中喹诺酮类药物是水产品残留的主要兽药之一,具有抗菌效果好、抗菌谱广等特点,在水产养殖中被大量应用防治疾病[1]。如果长期食用喹诺酮类的食品可造成慢性中毒,引起细菌耐药性等后果[2]。
从2015 年开始我国在动物源食品中禁止使用的喹诺酮类药物有4 种,分别是诺氟沙星、氧氟沙星、洛美沙星和培氟沙星4 种兽药盐类、酯类及其他制剂,国家标准《食品安全国家标准 食品中兽药最大残留限量》(GB 31650—2019)[3]中鱼(皮+肉)中恩诺沙星和环丙沙星之和残留限量为100 µg·kg-1。虽然针对水产养殖用兽药和非处方抗生素的使用有相应的控制和监管措施,但在养殖过程中仍会有过度使用导致药物在动物食品中富集,通过生物链进入人体内,从而对人体健康造成危害[4]。
目前,喹诺酮类药物常用检测方法主要有液相色谱法和超高效液相色谱-串联质谱(Ultra Performance Liquid Chromatography-Tandem Mass Spectrometry,UPLC-MS/MS)[5-7],而液质联用技术将超高效液相色谱和质谱的优点有效结合起来,具有专属性高、灵敏度高,适用于复杂基质中痕量组分分析优势,已成为当下兽药残留检测的主要手段。农业部1077号公告-1-2008[8]是目前检测水产品中喹诺酮类残留的UPLC-MS/MS 方法,但实际操作中发现该方法基质效应大,因此本实验在此方法基础上用固相萃取柱C18进行净化,有效改善基质效应的影响,保证结果的准确性。
1 材料与方法
1.1 材料与仪器
鲤鱼,购买于辽宁省营口市某农贸市场;诺氟沙星(纯度98.8%)、环丙沙星(纯度99.3%)、氧氟沙星(纯度99.8%)、培氟沙星(纯度99.8%)、洛美沙星(纯度99.7%)、恩诺沙星(纯度99.3%)、氘代诺氟沙星(诺氟沙星-D5,纯度99.3%)、氘代恩诺沙星(恩诺沙星-D5,纯度99.6%)和氘代环丙沙星(环丙沙星-D8,纯度99.6%),均购自ANPEL;甲醇、乙腈(色谱纯,德国Meker 公司);乙酸、甲酸(色谱纯,中国安谱有限公司);C18固相萃取柱(500 mg,6 mL,Aglient);试验用水均为超纯水。
Aglient 超高效液相色谱-质谱三重四极杆联用仪6460(配有EI 离子源);分析天平(感量0.01 g、0.000 01 g,瑞士Mettle Toledo 公司);涡旋振荡器(德国Heidolph);1 000 µL 移液枪(德国艾本德);冷冻离心机(美国Thermo Fisher Scientific);全自动氮吹浓缩仪N1(上海屹尧Preekem);Milli-Q Advamtage 超纯水器(美国Millipore)。
1.2 实验方法
1.2.1 标准溶液配制
准确称取培氟沙星、氧氟沙星、环丙沙星、诺氟沙星、洛美沙星和恩诺沙星对照品10 mg(精确至±0.000 01 g),分别置于10 mL 棕色容量瓶中,先加甲酸200 µL 使其完全溶解,再用甲醇定容,混匀配制成浓度为1 mg·mL-1的标准储备液。精确移取喹诺酮标准储备液适量,用甲醇稀释配制成0.1 µg·mL-1、1 µg·mL-1的混合标准工作液。
准确称取内标对照品诺氟沙星-D5、恩诺沙星-D5、环丙沙星-D85.0 mg、10.0 mg、2.5 mg(精确至±0.000 01 g),分别置于10 mL、10 mL、5 mL棕色容量瓶中,先加甲酸200 µL 使其完全溶解,再用甲醇定容,混匀配制成0.5 mg·mL-1、1.0 mg·mL-1、0.5 mg·mL-1同位素内标标准储备液。精密移取上述内标标准储备液适量,用甲醇稀释配成1.0 µg·mL-1的混合内标标准工作液。
1.2.2 样品前处理
称取匀浆样品5.0 g(准确至±0.02 g),置于50 mL塑料离心管中,准确加入50 µL 诺氟沙星-D5、恩诺沙星-D5、环丙沙星-D8混合内标标准工作液,涡旋混匀30 s,避光放置10 min,加入10 mL 1%乙酸乙腈,涡旋混合1 min,超声提取5 min,10 000 r·min-1,离心5 min,上清液转移到另一个50 mL 离心管中,向残渣中加入10 mL 1%乙酸乙腈重复提取1 次,合并两次提取液,于40 水浴中氮吹至近干,用5 mL 10%甲醇水溶解残渣,涡旋混匀备用。用6 mL 甲醇和6 mL 水依次活化净化柱C18,取5 mL 备用液上样,加水6 mL 淋洗,每次3 mL,弃去流出液,用6 mL 甲醇洗脱,将洗脱液在40 水浴中氮吹至近干,1 mL流动相定容;过0.22 µm有机微孔滤膜至进样瓶,LC-MS/MS 待测定。
1.2.3 标准曲线制备
采用空白样品所得溶液稀释标准溶液制得空白基质标准曲线。取空白样品按照“1.2.2”的方法处理,在经提取净化氮吹后的残余物中加入流动相制得空白基质,添加适量混合标准工作液制得浓度分别为0.01 µg·mL-1、0.02 µg·mL-1、0.05 µg·mL-1、0.10 µg·mL-1和0.20 µg·mL-1系列标准工作液,其中内标浓度为0.05 µg·mL-1,供超高效液相色谱-串联质谱法测定。以测得定量离子峰面积和内标峰面积比为纵坐标、质量浓度为横坐标,制得标准曲线回归方程和相关系数。
1.2.4 仪器条件
(1)色谱条件。色谱柱:Agilent Poroshell 120 EC-C18柱(100 mm×2.1 mm,2.7 µm);柱温:35 ,进样量:5.0 µL,流速:0.3 mL·min-1。流动相:0.1%甲酸水-甲醇(0.1%甲酸),梯度洗脱程序见表1。
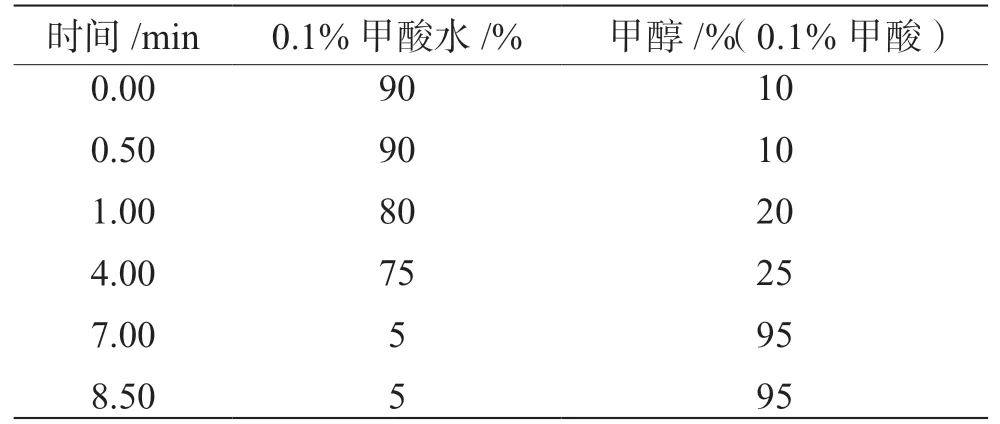
表1 液相色谱梯度洗脱程序表
(2)质谱MS 条件。负离子电喷雾ESI-多反应监测模式,扫描方式:负离子;毛细管电压:4 000 V;雾化气压力:40 psi;气流温度:325 ;鞘气流速:10 L·min-1。
1.2.5 样品的回收率和精密度
取空白鱼肉样品中添加5.0 µg·kg-1、10.0 µg·kg-1、50.0 µg·kg-1混合标准工作液,按“1.2.2”方法处理,进行加标回收实验,每个样品制备3 个平行样。
2 结果与分析
2.1 样品提取剂优化
本方法研究了水产品前处理中加入甲醇、乙腈、乙腈(1%甲酸)、乙腈(1%乙酸)对喹诺酮类药物的提取率的影响,其中乙腈(1%乙酸)的提取效果要大于乙腈(1%甲酸),这6 种喹诺酮对乙腈(1%乙酸)更敏感,切断化合物与蛋白质的结合,更容易溶于乙腈(1%乙酸),提取率会更高。因此,本实验方法采用乙腈(1%乙酸)作为提取剂。而单独的甲醇和乙腈提取率不佳,可能由于鱼肉基质复杂,导致喹诺酮与杂质复合沉淀,影响提取效果。
2.2 工作曲线线性范围
为了减少基质干扰,本实验采用空白基质配制标准曲线。结果表明,在0.01 ~0.20 µg·mL-1线性相关系数均大于0.99,线性关系良好,满足测定要求。线性回归方程及相关系数见表2。

表2 标准曲线的回归方程及相关系数
2.3 方法的回收率和精密度
在5.0 g 空白鲤鱼肉中添加低、中、高3 个浓度水平标准溶液,添加浓度分别为0.1 µg·mL-1喹诺酮混合标准溶液250 µL,1 µg·mL-1喹诺酮混合标准溶液50 µL,1 µg·mL-1喹诺酮混合标准溶液250 µL,添加浓度水平分别为5.0 µg·kg-1、10.0 µg·kg-1、50.0 µg·kg-1,添加的混合物内标浓度为0.05 µg·mL-1,每个添加水平做6 个平行,按照1.2.2 前处理的方法进行处理,测定喹诺酮峰响应值的相对标准偏差(Relative Standard Deviation,RSD)(%),计算方法的回收率和精密度。结果显示,3 个添加水平的平均回收率为89%~102%,精密度为3.9%~5.2%(表3),满足测定要求。
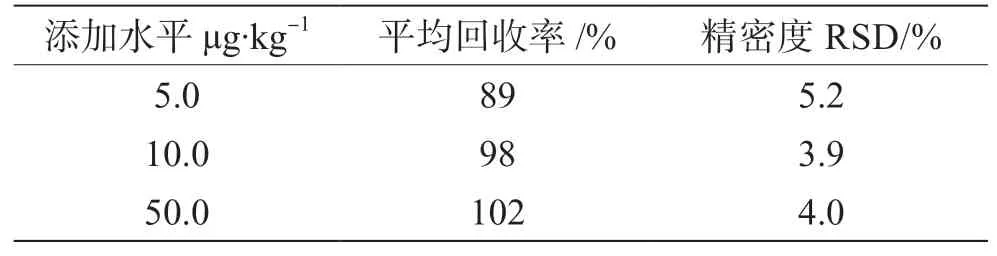
表3 方法回收率和精密度RSD 结果表(n=6)
3 结论
本方法建立了同时测定鲤鱼中6 种喹诺酮类兽药残留的超高效液相色谱-串联质谱分析方法。试样经酸化乙腈提取,固相萃取柱C18净化,能有效去除基质中大部分杂质,UPLC-MS/MS 分析检测。采用同位素内标法和空白基质匹配标准对检测结果进行校正,能有效改善基质效应的影响,保证检测结果的准确性。
