Cu13、Cu12Zr 和Cu12Zn 团簇上CO2 还原反应的密度泛函理论研究
2023-03-11李杰,李慧
李 杰,李 慧
(太原理工大学 材料科学与工程学院,山西 太原 030024)
化石燃料的大量使用导致CO2排放量不断增加,这是导致全球变暖的主要因素[1]。因此,降低大气中的CO2水平非常重要。三种主要的CO2减排策略分别是CO2排放截止、CO2储存和CO2使用[2,3]。其中,将CO2转化为增值化学品,尤其是甲醇,一直是该领域的研究热点[4]。
CO2是一种惰性分子,需要使用催化剂来激活解离和氢化等过程。Gautam 等[5]基于第一性原理研究CO2在Cu7、Cu13和Cu19簇上的活化和吸附。研究结果表明,CO2分子在团簇上的吸附强度呈以下趋势:Cu13>Cu19>Cu7。二十面体Cu13簇将充当CO2解离成CO 和原子氧的前体。Padama等[6]进行了基于密度泛函理论(DFT)的计算,以研究CO2及其解离物质(CO 和O)在Cu3团簇负载在Cu(111)表面上的相互作用。为了比较,对Cu(111)进行了类似的研究。团簇区域对CO 和O 的吸附强于Cu(111)表面。另一方面,CO2微弱地吸附在表面上。关于CO2在Cu(111)上解离,文献发现,团簇降低了活化势垒并提供了对解离物质的更稳定吸附。一些研究表明,通过在铜表面引入第二种金属可以提高催化活性[7]。Jo 等[8]基于密度泛函理论和微动力学建模方法模拟了数量为1、2、3 个Zn 原子掺杂在Cu(211)块体阶梯位置对催化性能(活化能垒、频率转向和速率常数)和电子结构的影响。研究表明,单个Zn 原子取代Cu(211)阶梯状位点Cu原子提高了对CO2还原反应的催化活性。Li 等[9]研究了二十面体的55 个Cu 原子和一个Zr 原子掺杂在Cu55团簇中。利用密度泛函理论(DFT)计算反应物和中间产物的吸附能以及相关的能垒。所有获得的结果表明,Zr 掺杂在Cu55团簇表面使其具有优异的CO2还原的催化能力,即使是在块状Cu 表面掺杂Zr 原子时也是如此。因此,Zr 原子掺杂是有效提高Cu 基催化剂CO2还原活性的有效方法[9,10]。
因此,为了比较Zn 和Zr 掺杂在Cu 基团簇中对CO2还原反应的作用,本章选取了一个具有13 个Cu 原子组成的二十面体团簇结构,并使用DFT 方法对其催化还原CO2过程进行了研究。Cu 团簇表面有一个掺杂位点,相应的团簇表示为Cu12Zn 和Cu12Zr 团簇。计算了反应物和产物在Cu13、Cu12Zr 和Cu12Zn 表面上的最稳定的吸附位点、活化能和反应能。计算结果表明,团簇对吸附物的吸附能力呈现以下趋势:Cu12Zr >Cu13>Cu12Zn,且Zn 和Zr 元素的加入降低了CO2还原反应的能垒。
1 计算方法和模型
本工作所有的计算都是基于密度泛函理论(DFT)并在Materials Studio 8.0 软件包下的Dmol3模块下完成[11,12]。广义梯度近似(GGA)结合Perdew-Burke-Ernzerhof (PBE)能量泛函用于计算[13,14]。DFT semi-core Pseudopotentials(DSPP)代替金属内层的电子,用双数值基加轨道极化函数(DNP)扩展价电子波函数[15]。分子间范德华力由Grimme 色散校正[16]。对于所建立的团簇结构优化,利用0.005 Ha(1 Ha=27.2114 eV)的拖尾效应提高计算精度。能量、最大力和最大位移的收敛分别设置为1 ×10-5Ha、2 × 10-3Ha/Å和5 × 10-3Å。在计算过程中,电子的自旋极化不受限制。通过上述方法优化了初态(IS)和终态(FS)的结构和能量。通过使用完全线性同步和二次同步变换(LSQ/QST)的方法,选择能量最低的配置,即最稳定的结构来寻找过渡态(TS)[17,18]。每个过渡态都进行了频率分析,过渡态构型中有且只有一个虚频。
如图1,构建了具有13 个原子的二十面体(IH)结构的准球形纳米团簇模型。Cu13团簇的表面原子的配位数均为6(CN=6)。用单个Zr 原子和Zn 原子分别替换位于Cu 团簇表面顶点上的一个Cu 原子,得到Cu12Zr 和Cu12Zn 团簇。橘黄色的Cu 原子,蓝色的是Zr 原子,紫色的是Zn 原子。计算的参数有偏析能SE、吸附能Eads、活化能Ea和反应能ΔE。
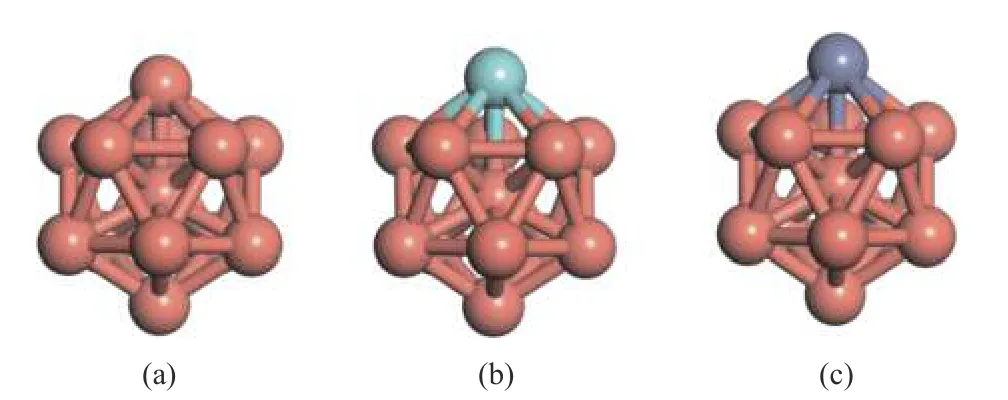
图1 三类团簇的结构模型示意图Figure 1 Structural model diagram of three types of clusters
偏析能SE值用以下公式计算:
式中,M是Zr 和Zn 原子掺杂在Cu13团簇,Mcore是Zr 和Zn 原子掺杂在Cu13团簇内部,E(Cu12M)和E(Cu12Mcore)是相应团簇的总能量。
催化剂表面吸附物的吸附能(Eads)使用下式计算:
式中,E[*adsorbate]是催化剂与吸附质的总能量,E[*]是纯的催化剂的能量,E[adsorbate]是吸附物在气相中的能量。根据这些定义,更负的Eads反映了团簇表面与吸附物更强的相互作用。其中,星号(*)代表吸附状态。
活化能(Ea)定义为反应过程中过渡态和初始态之间的能量差。反应能(ΔE)通常用来表示反应前后能量的变化。ΔE的正值表示吸热反应。Ea和ΔE基于以下公式计算:
式中,ETS、EIS和EFS分别是过渡态(TS)、初始态(IS)和终态(FS)的总能量。
2 结果与讨论
2.1 锌、锆掺杂在铜团簇的稳定性与团簇对中间体的吸附
Zn 和Zr 通常对催化反应表现出高催化活性。这些金属可能很昂贵,因此,通常使用Cu 基合金来减少昂贵金属的使用量,同时还能实现高催化活性。为了设计可行的合金催化剂,材料应该具有高催化活性,但也应该是稳定的。Cu 原子的半径为1.28 Å,Zn 原子的半径为1.39 Å,Zr 原子的半径为1.60 Å。已知具有较大半径的金属原子倾向于偏析到表面以减轻合金中的大应变[19]。在表1 中,计算了Zr、Zn 元素掺杂在Cu 团簇表面的偏析能(SE),Cu12Zr 的偏析能是-2.4 eV,Cu12Zn 的偏析能是-0.86 eV。越负的偏析能表明合金团簇在热力学上越稳定。
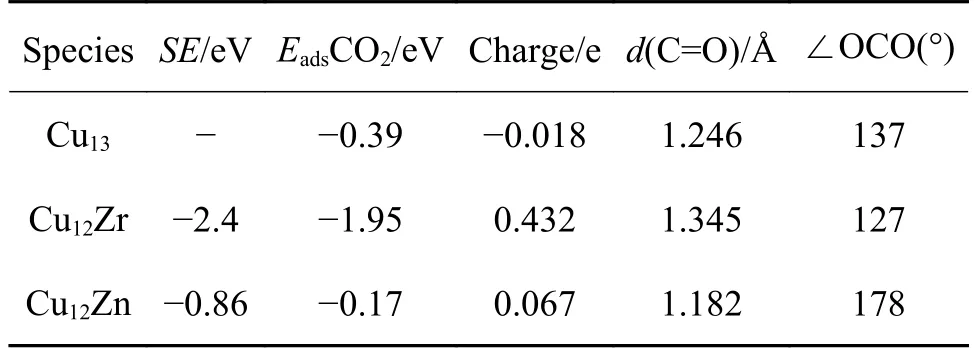
表1 Zr 和Zn 掺杂在Cu13 团簇表面的偏析能SE 和CO2 吸附参数Table 1 Segregation energy SE and CO2 adsorption parameters of Zr and Zn doping on the surface of Cu13 cluster
CO2吸附和活化是化学反应的关键步骤,旨在将其转化为其他有用的化学品。过渡金属簇的大比表面积与体积比可以激活高度热力学稳定的CO2分子。计算了锌、锆掺杂在Cu13团簇表面上对CO2分子吸附的影响,其吸附能值和几何参数见表1,吸附结构模型如图2 所示。
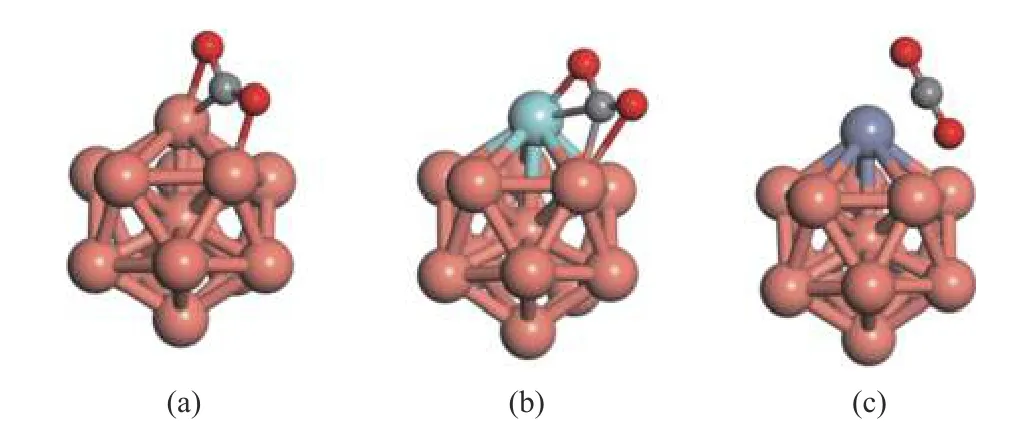
图2 (a)、(b)和(c)分别表示CO2 分子在Cu13、Cu12Zr 和Cu12Zn 簇上的吸附构型Figure 2 (a),(b) and (c) represent the adsorption configurations of CO2 molecules on Cu13,Cu12Zr and Cu12Zn clusters,respectively
CO2*在Cu13团簇中的吸附能为-0.39 eV,吸附后的CO2*的O-C-O 键角是137°,CO2*发生了弯曲属于化学吸附。Cu12Zr 团簇对CO2*的吸附能值为-1.95 eV,纯Cu13团簇表面掺杂一个Zr 原子提高了对CO2的吸附能力。CO2*分子的O-C-O 的键角是127°属于化学吸附。当Cu13团簇表面掺杂一个Zn 原子时,CO2*远离团簇表面,Cu12Zn 对CO2*的吸附减弱,吸附能值仅为-0.17 eV。O-C-O 的键角是178°,吸附构型是线性的且是物理吸附。同样有研究人员发现表面掺杂Zn 原子的Cu(211)和Cu(111)块体对CO2是弱吸附[8]。
此外,计算了CO*、HCOO*、COOH*、H*和O*在Cu13、Cu12Zr、Cu12Zn 团簇上的稳定吸附,吸附构型和吸附能(eV)如表2 所示。吸附能绝对值大小呈以下趋势:Cu12Zr >Cu13>Cu12Zn。Zr 原子掺杂在Cu13团簇表面增强对反应物的吸附能力,而Zn 原子掺杂在Cu13团簇表面降低了对反应物的吸附能力。
2.2 锌、锆掺杂在铜团簇表面对CO2 还原反应的研究
将CO2转化为任何有价值的化学产品的第一步是吸附和活化这种高度惰性的分子。计算了在Cu13、Cu12Zr 和Cu12Zn 团簇CO2还原反应活化能垒(Ea)和反应能(ΔE),列于表3 中。团簇表面上每个基本步骤的过渡态结构和势能图,如图3-图5所示。
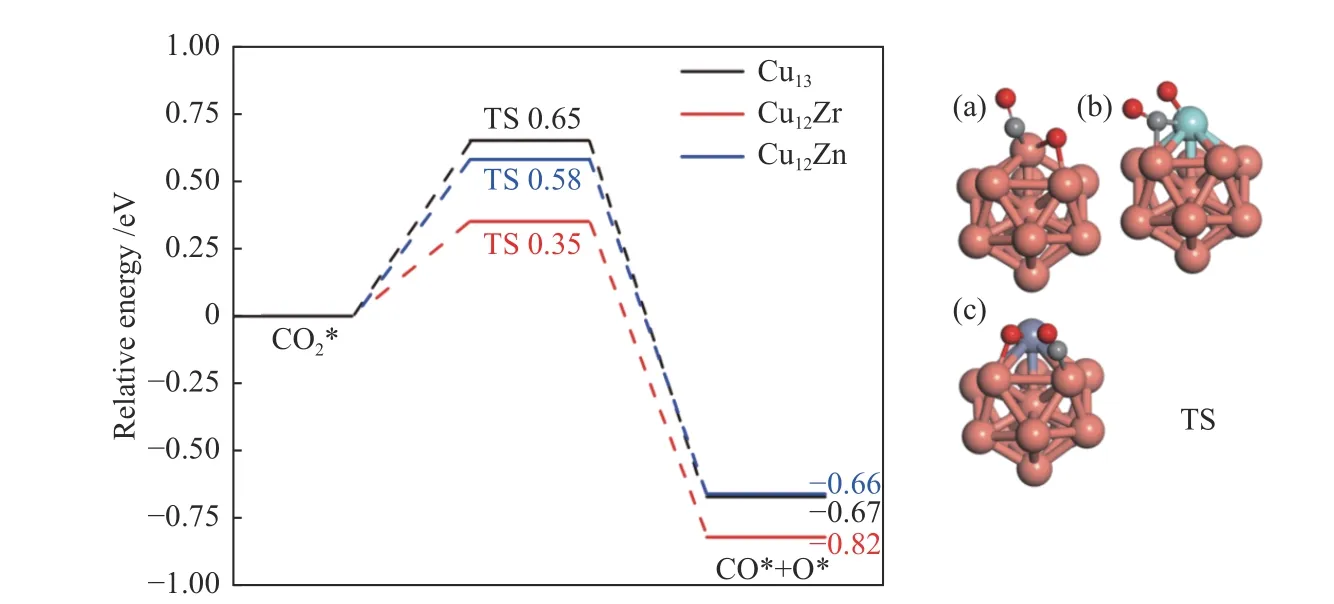
图3 CO2*解离成CO 和O 势能图及(a)Cu13、(b)Cu12Zr 和(c)Cu12Zn 团簇表面过渡态结构的对应描述C、O 和H 原子以灰色、红色和白色显示Figure 3 CO2* dissociation into CO and O potential energy diagrams and corresponding descriptions of the surface transition state structures of (a) Cu13,(b) Cu12Zr and (c) Cu12Zn clusters (C,O and H atoms are shown in grey,red and white)
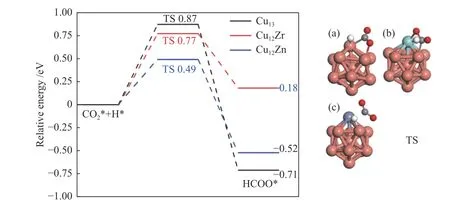
图4 CO2*加氢生成HCOO*势能图以及(a)Cu13、(b)Cu12Zr 和(c)Cu12Zn 团簇表面过渡态结构的相应描述C、O 和H 原子以灰色、红色和白色显示Figure 4 Hydrogenation of CO2* to HCOO* potential energy diagram and corresponding descriptions of the surface transition state structures of (a) Cu13,(b) Cu12Zr and (c) Cu12Zn clusters (C,O and H atoms are shown in grey,red and white)

图5 CO2*加氢生成COOH*势能图以及(a)Cu13、(b)Cu12Zr 和(c)Cu12Zn 团簇表面过渡态结构的相应描述C、O 和H 原子以灰色、红色和白色显示Figure 5 Hydrogenation of CO2* to COOH* potential energy diagram and corresponding descriptions of the surface transition state structures of (a) Cu13,(b) Cu12Zr and (c) Cu12Zn clusters (C,O and H atoms are shown in grey,red and white)
表3 Cu13、Cu12Zr 和Cu12Zn 团簇CO2 还原的活化能Ea 和反应能ΔETable 3 Activation energy Ea and reaction energy ΔE of CO2 reduction on Cu13,Cu12Zr and Cu12Zn clusters
CO2*在Cu13团簇和Cu12Zr 团簇上是化学吸附,而在Cu12Zn 团簇上是物理吸附。首先,CO2*可以直接解为CO*和O*。在过渡态中(图3)C-O键断裂,C-O 键的距离在Cu13、Cu12Zr 和Cu12Zn 团簇减小为1.67、2.65 和1.48 Å。该过程在Cu13、Cu12Zr和Cu12Zn 团簇上克服的能垒分别为0.65、0.35 和0.58 eV。相应的反应能为-0.67、-0.82 和-0.66 eV,该过程是放热反应。能垒呈以下趋势:Cu13>Cu12Zn >Cu12Zr。第二种掺杂元素Zr、Zn 的加入降低了CO2*解离为CO*的能垒。
同样,CO2*中的C 原子可以与氢结合生成HCOO*。在过渡态中(图4) H 原子向C 原子移动,H C 键距离在Cu13、Cu12Zr 和Cu12Zn 团簇减少为1.38、1.66 和2.59 Å。该过程在Cu13、Cu12Zr 和Cu12Zn 团簇上克服的能垒分别为0.87、0.77 和0.49 eV。相应的反应能为-0.71、0.18 和-0.52 eV。该过程在Cu13和Cu12Zn 团簇上是放热反应,而在Cu12Zr 团簇上是吸热反应。能垒呈以下趋势:Cu13>Cu12Zr >Cu12Zn。
CO2*中的O 原子可以与H 原子结合生成COOH*。在过渡态中(图5) H 原子向O 原子移动,O-H 键在Cu13、Cu12Zr 和Cu12Zn 团簇距离减少至1.66、1.49 和1.74 Å。该过程在Cu13、Cu12Zr 和Cu12Zn 团簇上克服的能垒分别为1.67、1.89 和0.99 eV。相应的反应能为0.41、0.63 和0.47 eV,该过程是放热反应。能垒趋势为:Cu12Zr >Cu13>Cu12Zn。Zr 元素的加入提高了纯Cu13团簇CO2*加氢生成COOH*的能垒,而Zn 元素的加入降低了纯Cu13团簇上CO2*加氢生成COOH*的能垒。
2.3 电子结构分析
综合以上分析可知,Cu13团簇中表面掺杂Zr 原子和Zn 原子会影响催化剂的催化性能。计算了Cu13、Cu12Zr 和Cu12Zn 团簇的Mulliken 电荷,以阐明其组成对CO2还原反应影响的微观原因,如图6 所示。
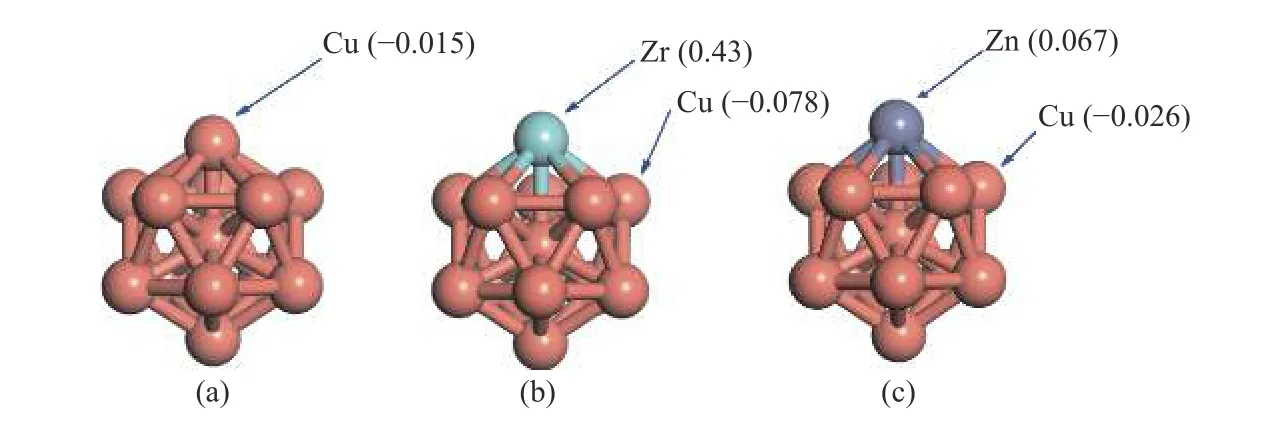
图6 (a)Cu13、(b)Cu12Zr 和(c)Cu12Zn 团簇表面原子的电荷分布Figure 6 Charge distribution of surface atoms of (a) Cu13,(b) Cu12Zr and (c) Cu12Zn clusters Negative values indicate electron gain,positive values indicate electron loss
电子转移和原子的电负性紧密相关,Cu、Zr和Zn 原子的电负性分别为1.90、1.33 和1.60。这说明,表面Cu 原子与Zn 和Zr 原子之间会发生电荷转移,Cu 原子得电子带负电荷而Zr 和Zn 原子分别失电子带正电荷。因此,可以得出结论,Zr和Zn 原子充当Cu 基团簇中的电子供体,而Cu 原子充当Cu 基团簇中的电子受体。在吸附的过程中,Cu12Zr 团簇的Zr 原子的电子转移量0.43 大于Cu12Zn 的团簇的Zn 原子的电子转移量0.067,因此,反应物和中间产物在Cu12Zr 团簇中的吸附能力是较高的。
此外计算了Cu13表面的Cu 原子、Cu12Zn 表面的Zn 原子和Cu12Zr 表面的Zr 原子的d电子轨道态密度图。由图7 比较Cu13、Cu12Zn 和Cu12Zr 团簇上原子d电子轨道分布可以看出,Zn 原子和Zr原子的d带中心下移,这归因于表面Zn 和Zr 原子之间电荷转移的程度不同,从而影响了其催化活性。

图7 Cu13 表面的Cu 原子(黑色)、Cu12Zn 表面的Zn 原子(蓝色)和Cu12Zr 表面的Zr 原子(红色)的d 电子轨道态密度图Figure 7 The d electron orbital density of states of Cu atoms(black) on the surface of Cu13,Zn atoms (blue) on the surface of Cu12Zn,and Zr atoms (red) on the surface of Cu12Zr,respectively (the dashed line at 0 eV represents the Fermi level)
3 结 论
相比于Cu13团簇,Cu12Zr 增强了对反应物的吸附能力,而Cu12Zn 降低了对反应物吸附能力。Zn和Zr 元素的加入降低了CO2还原反应的能垒,有利于CO2还原反应的进行。此外,Cu12Zr 团簇更倾向于把CO2还原为CO,而Cu12Zn 团簇更倾向于CO2还原为HCOO,掺杂元素种类的改变对CO2还原反应的路径有重要影响。
