均相催化CO2加氢制备MeOH的研究进展
2022-11-24康杏思陈琼遥
康杏思,陈琼遥,何 林
(1.中国科学院兰州化学物理研究所羰基合成与选择氧化国家重点实验室,甘肃 兰州 730000;2.中国科学院大学,北京 100049)
全球气候变暖正在对人类社会构成巨大威胁,而CO2是导致全球变暖的温室气体之一。气候变暖会造成冰川融化、海平面上升、极端气候频发、水资源匮乏以及生态环境破坏等一系列问题,给人类生产与生活造成不可逆转的影响[1]。2020年,全球与能源相关的CO2排放量高达315亿t,2021年排放量则上升到363亿t,且该数字仍在不断增长[2-3]。从资源利用的角度来看,CO2是丰富的C1资源,可通过非还原性转化生产纯碱、尿素、水杨酸和碳酸酯类产品或加压至超临界状态作为溶剂使用[4-6];也可通过还原性转化得到CO、HCOOH、MeOH、C2+产物等高附加值产品。其中,CO2加氢制备的MeOH,是生产甲醛、二甲醚、乙酸和烯烃的重要原料,同时也可用作溶剂、燃料和汽油添加剂,是实现CO2资源化利用的重要路径之一,目前被广泛研究[7-13]。
从分子结构和价态方面考虑,CO2是一个三原子线型结构分子,碳原子处于+4价,最高氧化态使其具有很高的热力学稳定性和动力学稳定性。因此,要实现CO2的转化通常需要很高的外界能量输入,或是高能量的底物分子,例如H2[14-17]、B-H[18-19]、Si-H[20-21]以及Py-H[22]还原剂。在这些还原剂中,以H2还原CO2制备MeOH的经济性最好,同时也最具挑战。目前,均相CO2加氢制备MeOH有3条主要路径:CO2直接加氢制备MeOH(Path A);两步法,先CO2加氢生成CO2衍生物(如碳酸盐、甲酸盐、氨基甲酸盐、甲酸、酯、脲),这些衍生物本身具有较高能量,因此可在相当温和的条件下加氢制备MeOH(Path B);从CO2出 发,先 加 氢 制 备HCOOH,HCOOH再歧化生成MeOH、CO2和H2O(Path C)。本文主要总结了以H2为还原剂,按3条路径,由CO2加氢制备MeOH的最新进展,重点关注催化体系的设计和加氢反应机理。
1 CO2直接加氢制备MeOH
CO2直接加氢制备MeOH的催化体系是[Ru3(CO)12]-KI,在8 MPa、n(CO2)/n(H2)=1/3、240℃的条件下反应3 h,MeOH周转的最大圈数(TON)为32.0[23]。在反应过程中,同时生成CO、CH4、C2H6等副产物。探究产物浓度随时间的变化发现,MeOH是CO进一步加氢的产物;继续延长时间,体系中CH4、C2H6浓度会进一步增加,而MeOH浓度呈现下降趋势。根据红外数据分析推测,[Ru3(CO)12]在低温下先生成阴离子[HRu3(CO)11]-和[Ru(CO)3I3]-,随着温度的升高,[HRu3(CO)11]-迅速转变成[H3Ru4(CO)12]-,当温度上升到240℃时,可观察到CO2还原活性物种[H2Ru4(CO)12]2-[24]。为抑制CH4、C2H6的 生 成,J.Klankermayer等[25]开 发 了 一 步法实现CO2制备MeOH的Ru基催化体系,结果如图1所示。
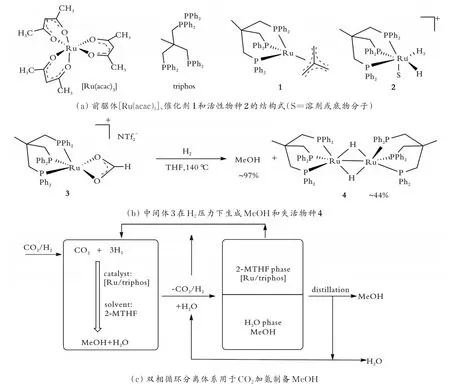
图1 一步法实现CO2制备MeOH的Ru基催化体系Fig.1 Ru-based catalytic system for the preparation of MeOH by CO2 was realized in one step
首先,在3.0 MPa、140℃的条件及甲基磺酸(MAS)的辅助下,采用催化剂[Ru(acac)3]/triphos和催化剂1(见图1(a)),考察了甲酸甲酯和甲酸乙酯的加氢性能,反应24 h,TON约为75.0。然后,将[Ru(acac)3]/triphos/MSA和催化剂1/HNTf2体系分别用于CO2加氢制备MeOH,同样反应24 h,TON分别为135.0和221.0。J.Klankermayer等[25]发现,酸能加速催化前驱体转化为Ru活性物种2的过程,从而明显提高催化活性;在体系中未检测到CO和CH4等副产物。进一步研究发现,在催化剂1/HNTf2体系中,中间体3是关键,进一步加氢生成MeOH,同时形成一部分失活物种4[26](见图1(b))。此 外,S.Wesselbaum等[26]设 计 了2-MTHF/H2O双相体系用于催化剂回收和循环(见图1(c))。结果表明,尽管反应体系能循环,但在循环4次后催化活性已经降低50%。
为提高CO2加氢制备MeOH的反应活性,J.Klankermayer等[27]对Ru催化体系进行了进一步改进。结果发现,催化剂5(见图2)的催化活性远高于催化剂1;在12.0 MPa、n(CO2)/n(H2)=1/3、120℃的条件下反应20 h,TON达到2 148.0,初始转化频率(TOF)为458 h-1。催化剂5的电子和位阻效应同催化剂1类似,但前者存在环己烷骨架,因此严格限制了Ru构型,增强了催化剂的稳定性,该体系在高碳醇/H2O双相体系中也表现出了很高的活性,水相中MeOH浓度达到2.5 mol/L。

图2 催化剂5用于CO2还原制备MeOH[27]Fig.2 Catalyst 5 tested in CO2 hydrogenation to MeOH
金属Ru价格较高,大规模工业化需要较高的成本,因此相关人员进行了3d金属用于CO2加氢制备MeOH的研究。M.Beller课题组将非贵金属[Co(acac)3]/triphos体系用于CO2直接加氢制备MeOH,该体系需要Lewis酸和Bronsted酸的辅助[28]。在8 MPa、n(CO2)/n(H2)=1/3、140℃的 条件下,反应24 h,TON达到31.0。在此基础上,将温度降低到100℃,同时提高HNTf2的物质的量和H2压力(7.0 MPa),TON可提高至50.0,然而反应48 h后催化剂完全失活。高分辨质谱和高压原位31P NMR分析结果表明,反应通过内层氢转移机理实现,与Ru(triphos)(TMM)体系制备MeOH类似[25]。为提高[Co/triphos]体系的催化活性,M.Beller课题组尝试筛选膦配体、Co盐和酸种类,但未获得催化活性更高的体系[29]。另外,该体系中的产物MeOH、H2O和CO会抑制催化剂活性。
2 CO2两步法加氢制备MeOH
由于CO2直接还原为MeOH存在巨大挑战,到目前为止,大多是在温和的条件下通过两步法合成MeOH。M.S.Sanford等[30]开发了由三催化剂体系串联完成CO2加氢制备MeOH的工艺(见图3(a))。

图3 三催化剂体系用于CO2还原制备MeOHFig.3 Three-catalysts system for the synthesis of MeOH from CO2
[RuCl(OAc)(PMe3)4]先将CO2还原为HCOOH,然后Sc(OTf)3将HCOOH酯化为甲酸甲酯,最后再由催化剂6完成酯还原制备MeOH的过程。在4.0 MPa、n(CO2)/n(H2)=1/3、135℃的条件下,由CO2制备甲酸甲酯过程的TON为40.0,但若是三种催化剂一锅法还原CO2制备MeOH,在相同条件下总TON仅为2.5。其主要原因是在高压CO2以及Sc(OTf)3存在的条件下,催化剂6迅速失活。为解决这个问题,M.S.Sanford等[30]设计了双管反应器(见图3(b)),将CO2制备甲酸甲酯的催化剂置于内管,催化剂6置于外管,原位生成的甲酸甲酯从内管挥发至外管中,从而提高了整个催化循环的效率,改进后总TON可提高到21.0。
最近,K.I.Goldberg课题组运用三催化剂体系还原CO2制备MeOH[31]时,为提高体系活性,对两步加氢所需的催化剂进行了筛选,最终优化出催化剂7、Sc(OTf)3和 催 化 剂8的 催 化 剂 组 合,在9 MPa、n(CO2)/n(H2)=1/8、155℃的条件下,反应40 h,该组合的TON为428.0(见图4)。在反应过程中,伴随着RWGS(逆水煤气变化)过程产生的CO与Ru催化剂作用形成无CO2加氢活性的物种Ru[P(CH3CH2PPh2)3](H)(CO)](OTf),而该物种限制了体系活性;体系中的四齿膦配体保证了Ru催化剂的稳定性,且Sc(OTf)3不会导致Ir-PCPtBu分解。因此,该催化剂体系并不需要设计双管体系即可得到可观的TON。

图4 三催化剂体系实现一锅法从CO2制备MeOHFig.4 Cascade catalysis through one-pot method for CO2 reduction to MeOH
D.F.Wass等[32]报道了二级胺辅助的络合物9(见图5)催化CO2加氢制备MeOH的方法。胺的种类对反应影响较大,在三级胺存在下体系无活性,使用的二甲胺所对应的产物主要为甲酰胺,经筛选发现二异丙胺效果最好。在4.0 MPa、n(CO2)/n(H2)=1/3、100℃的条件下,该体系的最高TON达到8 900.0,降 低 催 化 剂 浓 度 到nmol级,TOF达 到4 500 h-1。
除上述膦配体外,N.P.Rath课题组发现PTA(1,3,5-三氮杂-7-磷杂金刚烷)络合的Ru络合物10(见图5)能在非常温和的条件下(0.4 MPa、n(CO2)/n(H2)=1/3、60℃)还原CO2制备MeOH[33],反应24 h后的TON达4 752.0;络合物11在同样条件下的TON也可以达1 000.0以上。Rh-PTA络合物是目前报道的第一个Rh基均相催化CO2制备MeOH。这一体系需要酸作助剂,且MSA和HNTf2的效果最好,该课题组推测酸能促进体系中Ru-H和Rh-H的 形成。

图5 Ru、Rh络合物体系用于温和条件下CO2加氢制备MeOHFig.5 Ru and Rh complexes examined for CO2 hydrogenation to MeOH under mild conditions
M.S.Sanford等[34]采用催化剂12,在二甲胺辅助下,将CO2经甲酰胺中间体还原制备了MeOH。该体系的关键在于二甲胺能捕捉CO2形成DMC。首先,将DMC加氢制备MeOH作为模型反应优化反应条件,在反应温度为155℃、K3PO4的摩尔分数为50%、THF作溶剂的催化条件下,MeOH的TON达到19.0。图6为催化剂12在二甲胺辅助下经甲酰胺中间体还原制备MeOH。由图6(a)可知,DMC加氢制备MeOH有两种加氢路径:DMC直接加氢生成DMF,然后DMF进一步加氢制备MeOH(path I);DMC先 分 解 为CO2,然 后CO2被 还 原 为HCOOH,最后经胺化生成DMF(path II)。由图6(b)可知,DMC最难被还原。M.S.Sanford等[34]发现,在95℃的条件下,DMC经path I可被还原为MeOH,因此认为经path II更合理。M.S.Sanford等[34]还尝试了分步法制备MeOH。首先,在5.3 MPa、n(CO2)/n(H2)=2.5/50、95℃的 条 件 下 制 备DMF,再将温度升高到155℃,使DMF还原为MeOH,该 过 程CO2转 化 率 高 达96%,TON为550.0。体系中胺有两方面的作用,一是与CO2反应生成DMC,其次是与HCOOH作用产生DMF。

图6 催化剂12在二甲胺辅助下经甲酰胺中间体还原制备MeOHFig.6 Amine assisted sequential methanol synthesis from CO2 via N-formylation in the presence of catalyst 12
D.Milstein等[35]发展了在碱性条件下乙醇胺捕捉CO2形成氨基甲酸酯,氨基甲酸酯继续加氢制备MeOH的催化体系,CO2压力可低至0.1~0.3 MPa(见图7)。对两步过程进行了探讨:第一步,在150℃的条件下,Cs2CO3催化醇胺与CO2作用生成氨基甲酸酯,醇胺上氮的α位上取代基位阻越大越有利;第二步,在H2分压为6.000 MPa的条件下,催化剂13催化氨基甲酸酯加氢,在Ru、tBuOK的摩尔分数均为1%、135℃的条件下反应19 h,氨基甲酸酯转化率为92%。基于以上结果,尝试了不分离中间体,用催化剂13、Cs2CO3共同催化CO2加氢制备MeOH。结果表明,在CO2分压为0.100 MPa的条件下,最终MeOH产率达53%。该体系的优势在于常压CO2能被还原为MeOH。

图7 乙醇胺衍生物捕捉CO2经氨基甲酸酯制MeOHFig.7 Synthesis of methanol via carbamate using aminoethanol derivatives to capture CO2
G.K.S.Prakash课 题 组 采 用M.S.Sanford等[34]的胺捕捉CO2策略,利用易回收的PEHA捕捉CO2,尝试了一系列Ru-PNP催化剂在PEHA存在下的加氢活性[36]。结果表明,在7.5 MPa、n(CO2)/n(H2)=1/3、155℃的条件下,催化体系催化剂12/PEHA制备MeOH的TON达 到1 060.0,此 时CO2转 化 率 为23%。将反应体系中的MeOH、H2O和溶剂通过蒸馏分离,催化剂直接用于下一次循环。循环5次后,催化剂活性仍保持初始活性的75%,催化剂的总TON达1 850.0。将n(CO2)/n(H2)从1/3改 为1/9,温度降到145℃,反应200 h后,MeOH产率可提高到65%,总TON为1 200.0,初始TOF为70 h-1。该体系的CO2可直接从空气中捕获。尽管CO2质量分数较低(400 μg/g),MeOH产率仍可达61%,延长反应时间,产率可提高到79%。这项研究为直接将空气作CO2来源合成MeOH迈出了重要的一步。
G.K.S.Prakash课题组针对从CO2捕集到制取MeOH的串联过程,编制了完全可循环回收且高效的“工艺包”(见图8)[37]。首先,测试各种胺的水溶液 对CO2分 压 为0.007 MPa的 吸 收 能 力,4 h后PEHA水溶液中CO2的吸收量为11 mmol/g,折合成n(CO2)/n(N)为0.43。虽然工业上常用的乙醇胺对CO2吸收量更大(n(CO2)/n(N)为0.71),且相比于PEHA的价格($105),乙醇胺价格更低廉($35),但CO2乙醇胺水溶液中未检测到MeOH,仅检测到甲酸盐和甲酰胺。将2-MTHF和催化剂12加入水溶液中形成两相体系,再充入分压为7.000 MPa的H2,145℃下反应72 h,MeOH产率为47%,TON达到520.0。提高n(S)/n(C)到200,H2分压升高到8.000 MPa,MeOH产率可达95%,反应结束后只需简单蒸馏分离MeOH。采用这一策略,催化剂12和PEHA能循环3次且维持MeOH产率为87%,到第4次循环时活性只下降5%。这项研究再次验证有望实现从空气或尾气中捕捉CO2来制备MeOH。

图8 催化剂12/PEHA双相体系一锅法实现CO2捕集和还原制备MeOHFig.8 One-pot two-step CO2 capture and hydrogenation to methanol by catalyst 12/PEHA catalysis using a biphasic setup
G.K.S.Prakash课题组对胺辅助Ru PNP催化还原CO2制备MeOH进行了系统的机理研究[38],探索了Ru pincer配体的电性、位阻的改变以及胺的种类对反应活性的影响(见图9),并借助原位1H、31P、13C-NMR和ATR-IR光谱等手段解释了实验结果。尽管大部分[RuHClPNPR(CO)](R=Ph/iPr/Cy/tBu)能催化CO2制备MeOH,但仅催化剂12表现出较高活性。另外,只有双氮类且不具有三级胺结构的胺才能促进MeOH的生成。体系中能检测到无活性的双羰基物种[RuHPNPPh(CO)2]+,这一物种经解离一个羰基后可转变为活性物种。延长反应时间至10 d,发现催化剂12仍保持很高的活性,在7.5 MPa((n(CO2)/n(H2)=1/1)、145℃的条件下,TON最高可达9 900.0。
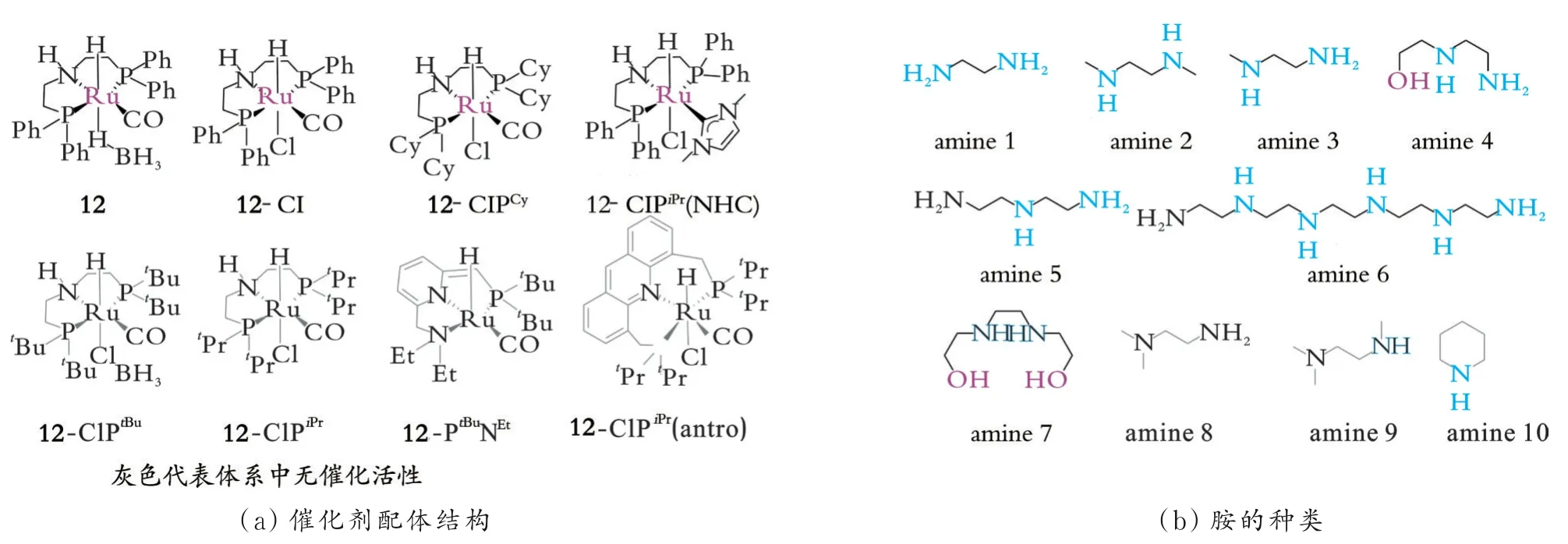
图9 催化剂配体结构和胺种类对MeOH产率的影响Fig.9 Effect of catalyst molecular structures and types of amine
Y.Kayaki课题组也采用同样的策略,利用PEIs(聚(乙烯亚胺))捕捉CO2,进一步将其还原为MeOH[39]。结果表明,与PEHA相比,PEIs吸收CO2需要更高的温度(100℃),但催化剂12能高效地将PEIs对应的甲酰胺还原为MeOH,在H2分压为5.000 MPa、155℃的条件下,MeOH产率高达94%。在该催化条件下,CO2加氢制备MeOH的总TON为599.0。
G.K.S.Prakash等[40]尝试利用固载化的胺(SSAs)捕捉CO2,并完成后续的氢化过程合成MeOH。结果表明,采用物理浸渍法、共价键法和交联法合成的8种SSAs,均表现出CO2加氢制备MeOH的活性,且通过简单过滤就能回收产物;通过浸渍法制备的胺化合物14(见图10)活性最好(反应40 h,TON为520.0)。核磁结果显示,反应过程中有胺的浸出,浸出率约为18%。相比之下,通过共价键吸附的固载化化合物15不存在胺浸出问题,循环3次后,MeOH产率基本不变,但在第3次循环时SSA捕捉CO2的能力降低50%。

图10 SSAs捕捉CO2再经催化剂12还原为MeOHFig.10 SSAs used in catalyzed 12 CO2 capture and hydrogenation
R.Sen等[41]报道了碱金属氢氧化物吸收CO2并将其还原为MeOH的反应(见图11)。结果表明,乙二醇在氢氧化物存在的条件下脱去质子得到醇盐,醇盐定量捕获CO2形成相应的烷基碳酸盐;在100~140℃的条件下,催化剂12能将烷基碳酸盐高效还原为MeOH,在H2分压为7.000 MPa、反应20 h的条件下,TON达到200.0。另外,该体系能直接吸收空气中的CO2,并将其全部转化为MeOH。但是,该体系中的乙二醇在碱性环境中脱氢生成碳酸盐,造成碱的消耗,因此每次循环时需要重新补充氢氧化物。该体系的TON(200.0)远低于胺辅助的Ru催化体系的TON(9 900.0)。
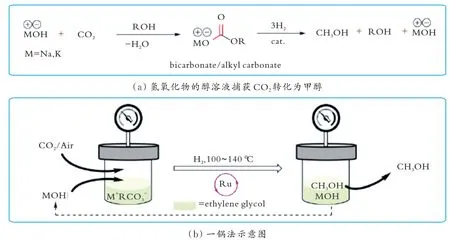
图11 利用碱金属氢氧化物的乙二醇溶液吸收CO2实现催化剂12催化CO2制备MeOHFig.11 Integrated CO2 scavenging using alkali hydroxide with ethylene glycol and subsequent catalyst 12 catalyzed CO2 to MeOH
W.Li等[42]采用催化剂16(见图12),通过一锅法将CO2还原为MeOH,催化剂16是一个高效内酯和酯类加氢催化剂[43]。该体系将二甲胺作为CO2捕获剂。在CO2分压为0.250 MPa、H2分压为5.000 MPa、170℃的条件下,将CO2通过甲酰胺中间体加氢制备了MeOH,反应72 h后总TON达2 100.0。提高CO2分压到0.500 MPa,会严重抑制催化剂活性,W.Li等[42]推测是由于CO2与配体上的氮原子作用造成的[44-45],但并未对失活机制做进一步探究。

图12 催化剂16用于CO2加氢制备MeOHFig.12 The catalyst 16 used in CO2 hydrogenation to MeOH
Fe与金属Ru处于同一主族,性质具有一定的相似性。因此,研究人员也将Fe用于催化CO2加氢还原过程。A.J.L.Pombeiro等[46]发现三齿氮配体络合的Fe催化剂17(见图13)在温和条件(7.5 MPa、n(CO2)/n(H2)=1/3、80℃)下反应24 h,能高效催化CO2制 备MeOH,总TON达2 283.0,折 合TOF为95 h-1。吡唑基上氮原子能促进H2异裂,添加PEHA能明显提高TON,这意味着反应过程很可能经过甲酰类中间体;不加PEHA,MeOH产率从45%降到36%,同时碳利用率也从86%降到41%。Fe基催化剂虽然表现出很高的活性,但在反应24 h后催化剂活性降低50%。对体系的稳定性和反应机理值得进一步探究。

图13 Fe催化剂17用于CO2加氢MeOHFig.13 Fe-based catalyst 17 tested in CO2 hydrogenation to MeOH
3 HCOOH歧化制备MeOH
HCOOH歧化制备MeOH策略:首先,CO2加氢制备HCOOH,再歧化生成MeOH、CO2和H2O。2013年,A.J.M.Miller等[47]将一种“半三 明 治”催化剂18用于HCOOH加氢制备MeOH(见图14)。在催化剂18质量分数为20 μg/g、HCOOH浓度为12 mol/L、80℃的条件下反应24 h,MeOH选择性仅为7%,总产率为2%,大部分HCOOH分解为CO2和H2。

图14 催化剂18用于HCOOH歧化制备MeOHFig.14 The catalyst 18 used in disproportionation of HCOOH to MeOH
T.Cantat课题组将[Ru(COD)(methylallyl)2]/triphos催化体系用于HCOOH加氢制备MeOH[48]。与文献[25]报道的Ru催化CO2间接加氢制备MeOH体系类似,在150℃条件下MeOH产率为50%。结果表明,连续氢转移是生成MeOH的路径之一,且HCOOH分解生成的H2有助于氢转移步骤中甲醛加氢制备MeOH(见图15)。

图15 HCOOH歧化制备MeOH的路径Fig.15 Proposed pathways for the disproportionation of formic acid to MeOH
G.Laurenczy和Y.Himeda课题组提出了Ir催化剂19在常温下间接还原CO2制备MeOH的方法(见图16)。CO2先还原为HCOOH,再经歧化生成MeOH[49-50]。13C-NMR结 果 表 明,MeOH是 由HCOOH转化而非CO2直接还原的结果[51]。

图16 Ir催化剂19用于HCOOH加氢制备MeOHFig.16 Disproportionation HCOOH to MeOH by Ir complex catalyst 19
虽然催化剂19可同时完成CO2还原到HCOOH,再经歧化制备MeOH这两步过程,但均是对每一步分别研究,并未有以CO2为底物直接制备MeOH的结果。R.Kanega等[52]在Ir催化体系上取得了进展(见图17(a))。由于单核M-H更倾向于和CO2作用形成甲酸根中间物种,再与溶剂中H2O发生配体交换得到HCOOH,R.Kanega等[52]推测多核金属M-H物种可在分子内发生连续负氢转移,从而可在催化剂分子内直接形成MeOH。体系中应避免H2O的存在,因此采用了气固相装置。结果表明,采用多核Ir催化剂在气固相装置及温和条件下可实现CO2直接加氢制备MeOH。同时,因为该装置本身的特点,产物和催化剂只需通过泄压即可分离。在4.0 MPa、n(CO2)/n(H2)=1/3、60℃的条件下循环5次后,总TON为113.0,且1H-NMR结果显示该催化剂并无明显变化。
R.Kanega等[52]还对常规水相体系和气固相体系的不同机理进行了推测(见图17(b))。首先,两种反应体系均是在H2氛围中产生Ir-H物种(step i);然后,CO2插入Ir-H物种,得到甲酸根中间体B(step ii)。在进一步进行的加氢反应过程中,两种体系表现出不一样的加氢行为。在无水参与的气固相体系中,分子内的一个Ir-H转移到临近的Ir-OOCH上,得到络合物C(step iii),并通过类似于负氢转移的过程得到Ir的甲氧基络合物D(step iv),二维核磁表征结果佐证了体系中络合物D的存在。最后,催化过程完成第三次分子内负氢转移得到MeOH(step v)。在水相体系中,因H2O的存在,甲酸根中间体B与H2O发生配体交换得到HCOOH(step vi)。在同一反应条件下,HCOOH被Ir络合物分解为CO2和H2O。因此,该研究阐释了在气固相反应体系中多核Ir络合物能在60℃的低温下催化CO2加氢制备MeOH,在均相体系中检测到少量HCOOH。

图17 多核Ir体系用于CO2加氢制备MeOHFig.17 Multinuclear Ir complex catalyst for CO2 hydrogenation to MeOH
4 总结与展望
大气中CO2质量分数的不断升高,是导致气候变暖的主要原因,因此应从源头上降低其释放量,并通过资源利用的方式利用已产生的CO2。总结了均相体系中经3种不同的还原路径将H2还原CO2制备MeOH的研究进展。其中,由CO2直接制备MeOH的路径,目前面临的问题是需要较高的温度、压力,这对反应装置的设计是巨大的挑战;对两步法经CO2衍生物制备MeOH的路径,需要考虑体系中前后两步催化剂的兼容性问题,尽管目前报道的TON数据比较可观,但都是10-6级的催化剂,且反应时间长,这与实际生产有很大的距离;CO2先还原为HCOOH,HCOOH再歧化得到MeOH的路径。该路径中MeOH选择性比前两个路径低。这是因为大多数CO2加氢制备HCOOH的催化剂,同样能催化HCOOH分解为CO2和H2[53]。因此,催化体系的设计尤为重要,研究均相催化CO2还原制备MeOH的意义在于均相体系活性位点的确定性有利于催化机理的研究,这将有助于启发和促进更高效的低温多相催化体系的开发。
在所有还原CO2的还原剂中,H2作为还原剂的反应的原子经济性最高。未来若能实现大规模光解水制氢或利用绿色电力的电解水制氢,将大幅提升CO2的资源化利用效率。迄今为止,尽管已有大量关于CO2还原为MeOH的研究[9,17],但要实现CO2制备MeOH的大规模产业化,仍有许多问题需要解决。
(1)均相催化体系普遍存在催化剂稳定性和难回收等问题,因此工业上更倾向于使用多相催化剂[54-55]。目前,CO2加氢制备MeOH的多相体系存在反应活性差、选择性低等问题。因此,开发更加低温高效的多相催化体系依然是研究的重点内容。
(2)目前,均相分子型催化剂的研究主要集中于Ru基络合物,对3d金属络合物的研究相对较少。加强3d金属络合物的研究,有望降低CO2制备MeOH的工业成本。
(3)大多数对CO2加氢制备MeOH的研究都集中在开发高活性和稳定催化剂的催化体系上,但对于在对应的催化反应条件下催化剂的动态行为、体系的活性位点和反应机理的研究相对不够深入。因此,如果能够在反应条件下对催化剂原位结构变化进行研究,并结合理论模型理清活性位点、构-效关系以及反应机理,则可加快CO2制备MeOH的工业化进程。
(4)目前,虽然可在常压下由CO2制备MeOH,但反应效率低,大多需要高纯度、高压CO2原料,而且从大气中分离CO2需要耗费巨大的能量,利用工业尾气或空气中的低浓度CO2原料仍是需要解决的问题。
最后,希望研究人员迎接CO2加氢制备MeOH存在的挑战,提高CO2资源的利用率,为解决全球变暖化问题做出贡献。
