肾消耗病
2022-11-22梁丹丹陈文萃曾彩虹
梁丹丹 陈文萃 曾彩虹
病例摘要
病史男性患者,14岁,因“发现双肾囊肿,血清肌酐(SCr)升高近2年”于2021-10-18入院。
患者2019年11月因上呼吸道感染于当地医院就诊,检查发现双肾囊肿,SCr 127 μmol/L,未予特殊治疗。2020年3月复查尿检阴性,SCr仍偏高。2020年5月起服用百令胶囊、罗盖全及碳酸钙D3治疗。2020年10月当地查甲状旁腺素115 pg/mL,胸部CT示骨质密度不均,双肾B超示双肾多发性囊肿,双肾皮质多发点状强回声,头颅MR平扫示双侧半卵圆中心、双侧侧脑室体部旁、双侧皮质脊髓束走形区、左侧扣带回T2/FLAIR信号偏高,继续以补钙保肾治疗。此后多次复查SCr缓慢上升,最高达294 μmol/L,血红蛋白(Hb)99~108 g/L。2021-09-26于国家肾脏疾病临床医学研究中心就诊后收住入院。病程中精神较差,易疲劳,头晕、偶发视物旋转,易口渴。无咳嗽、咳痰,无胸闷、气短,无关节痛、频发口腔溃疡、皮疹,无视物模糊,无头痛,无腹胀、腹痛、便血。大便、排尿正常。
家族史父母健在,父亲体检肾脏超声提示单个肾囊肿,肾功能正常,母亲正常,父母亲为非近亲结婚。家族中无传染病家族史,无其他遗传性疾病家族史。
既往史、个人史无特殊。
体格检查体温36.8 ℃,脉搏75次/min,呼吸17次/min,血压138/60 mmHg,身高165 cm,体重36.5 kg,体质量指数13.4 kg/m2;神清,体形消瘦,咽不红,扁桃体无肿大,心、肺、腹部未见明显异常,双下肢无水肿。肌力正常。
实验室检查
血常规 Hb 91g/L,红细胞计数3.0×1012/L,白细胞计数4.32×109/L,血小板239×109/L,网织红细胞1.11%。
尿液 尿常规蛋白阴性,红细胞计数5.6/μL。尿蛋白定量0.44 g/d。24 h尿电解质(尿量1 530 mL):尿磷6.0 mmol/L,尿钠33.20 mmol/L,尿钙0.16 mmol/L,尿钾16.0 mmol/L,尿氯24.7 mmol/L。尿NAG、C3和α2-M正常,RBP 5.11 mg/L(正常值<0.5 mg/L)。尿酸化功能:pH 5.20,HCO3-29.00 mmol/L,TA 23.00 mmol/L,NH4+22.00 mmol/L,NAC 30.00 mmol/L,NH4+/TA 0.96。
血生化 谷丙转氨酶10 U/L,谷草转氨酶21 U/L,SCr 276.7 μmol/L,尿酸459 μmol/L,白蛋白44.2 g/L,球蛋白26.5 g/L,尿素氮16.2 mmol/L,磷2.01 mmol/L,钙2.25 mmol/L,氯108.0 mmol/L,钠146.7 mmol/L,钾3.78 mmol/L,总二氧化碳23 mmol/L,总胆固醇4.38 mmol/L,三酰甘油1.08 mmol/L;凝血功能正常。甲状腺功能正常。糖化血红蛋白5.3%。
其他ANA、A-dsDNA、抗核抗体谱阴性。IgG 12.7 g/L,IgA 2.11 g/L,IgM 0.480 g/L,IgE 36.4 IU/mL,抗链球菌溶血素O抗体、类风湿因子正常,补体C3 0.593 g/L,C4 0.172 g/L;PR3-ANCA、MPO-ANCA、抗GBM抗体阴性;免疫固定电泳图谱、抗磷脂酶A2受体抗体阴性;直接抗人球蛋白试验、间接抗人球蛋白试验阴性;血清叶酸13.99 ng/mL;血清维生素B12 280.00 pmol/L;红细胞碎片检查阴性。肝纤维化四项检Ⅲ型前胶原N端肽(PⅢNP)116.40 ng/mL(正常值0~30 ng/mL)。传染病四项阴性。甲状腺功能正常。
眼科检查视乳头边界清,黄斑区和视网膜正常,视网膜血管动静脉比例2∶3,双侧眼前节(-)。
影像学检查双肾B超:左肾102 mm×42 mm×51 mm、右肾101 mm×41 mm×50 mm,皮质回声增强,皮髓界限不清,双肾内见多个类圆形无回声区,左肾较大的约6 mm×6 mm,右肾较大的约6 mm×7 mm,部分囊周伴钙化。消化系统、颈部血管超、双侧颈部血管、甲状腺及甲状旁腺超声均未见异常。心脏超声:轻-中度三尖瓣反流。心电图:窦性心律;左心室高电压。
肾活检病理
光镜 皮髓质肾组织3条。41个肾小球中3个球性废弃,数个球发育欠佳、足细胞游离在囊腔内。余肾小球正切体积增大,节段系膜区轻度增宽,多个球毛细血管袢皱缩、开放欠佳、囊腔相对扩张,见无小管肾小球,数个球伴囊周纤维化(图1A),囊壁增厚分层。PASM-Masson:阴性。肾小管间质重度慢性病变,斑片状肾小管萎缩、基膜增厚分层,多处肾小管扩张、呈囊性改变(图1B、C),管腔内见少量蛋白管型、多处蛋白管型裸露于间质中,间质纤维化+++,大量炎细胞浸润,以单个核细胞为主,亦见灶性浆细胞。动脉未见明确病变。
免疫荧光 冰冻切片荧光染色示肾小球11个,其中3个硬化球。C3+,颗粒状、球性或节段沉积于肾小球系膜区。IgG、IgA、IgM、C1q、Fibrin、κ轻链、λ轻链阴性。
电镜 肾小球节段系膜区增宽,系膜区未见电子致密物分布。肾小球毛细血管袢开放好,偶见内皮下疏松,数处袢基膜扭曲、皱缩,基膜厚380~610 nm,基膜致密层无分层撕裂。肾小球毛细血管袢基膜内皮下、上皮侧未见电子致密物分布。肾小球足细胞足突偶见节段融合。观察的部分肾小管基膜增厚、分层(图1D)。
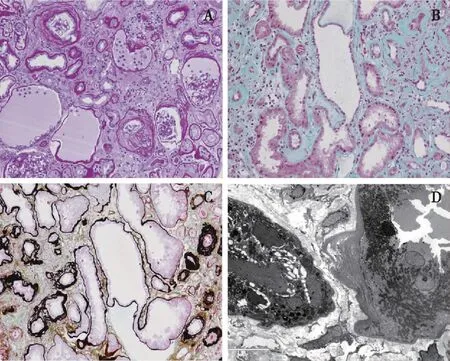
图1 A:多个肾小球毛细血管袢皱缩、开放欠佳、球囊腔相对扩张,囊腔内见游离的足细胞,囊壁增厚、分层;周围肾小管萎缩、基膜增厚,间质弥漫增宽、纤维化,较多单个核细胞浸润(PAS,×200);B:肾小管管腔呈囊样扩张,周围间质纤维化伴炎细胞浸润(Masson三色,×200); C:灶性肾小管萎缩、基膜扭曲增厚、分层,部分肾小管囊样扩张(PASM-Masson,×200);D:电镜下观察到肾小管基膜明显增厚分层(EM)
病理诊断慢性肾小管间质肾炎伴肾小管囊样扩张。
基因检测经二代基因测序发现该患者2号染色体上NPHP1基因存在纯合缺失突变,该突变导致NPHP1基因一段136 kb大小的碱基序列缺失,根据《美国医学遗传学与基因组学学会指南(2019年)》确定为致病性变异。其父亲和母亲均为杂合子。
讨 论
本例患者为青少年男性,病程2年,系偶然检查发现双肾囊肿、慢性肾功能不全,否认肾脏病家族史,肾外表现为易烦渴,发育尚正常。实验室检查示少量蛋白尿、SCr及尿液RBP升高,无镜下血尿,正细胞正色素性贫血,胸部CT示骨质不均。肾脏病理光镜表现为肾小管间质重度慢性病变,肾小管基膜增厚分层,部分肾小管囊样扩张,间质纤维化及炎细胞浸润,多个肾小球缺血皱缩,免疫荧光仅见少量C3沉积于肾小球,电镜下见肾小管基膜增厚、分层,肾小球病变不明显。
肾囊肿并不罕见,可由多种获得性、遗传性和发育异常性病因所导致。但对于青少年双肾多发囊肿,且肾脏病理表现为慢性肾小管间质肾炎伴多处肾小管囊性变,则需要对以下几种常见的肾囊肿性疾病进行鉴别。这些疾病多为遗传性疾病,主要包括(1)多囊肾:该病以常染色体显性遗传常见,即常染色体显性遗传多囊肾病(ADPKD),主要的致病基因是PKD1和PKD2,少数表现为常染色体隐性遗传;可累及全身多个脏器,患者多在成年后才出现双侧肾囊肿,常伴肝囊肿,肾脏损害临床表现为腰痛、腹痛、镜下或肉眼血尿、高血压、肾功能不全等,约半数患者在60岁时进展至终末期肾病(ESRD)[1]。该例患者无肝囊肿,父母双肾超声未见典型多囊肾改变,不支持ADPKD。(2)肾消耗病(NPHP):又称肾单位肾痨,是一种常见的导致儿童ESRD的遗传性肾病,主要表现为肾脏浓缩功能下降、慢性肾小管间质性肾炎、囊性肾病,30岁之前往往已进展至ESRD,最常见的导致NPHP的基因是NPHP1基因[2]。(3)常染色体显性遗传肾小管间质疾病(ADTKD):是一组由多种基因突变导致的遗传性疾病,包括UMOD、MUC1、HNF1B、REN[3],以及SEC61A1[4]。ADTKD临床表现为缓慢进展的肾功能下降、无或少量蛋白尿、病程早期通常无高血压,同时家族史阳性[3],部分患者会表现为高尿酸和早发痛风,影像学检查可见肾囊肿,ADTKD患者多在50岁以后才进展至ESRD;肾活检病理也表现为肾小管间质慢性病变,UMOD基因相关的ADTKD电镜下还可见远端肾小管上皮细胞内扩张的内质网以及尿调节素蛋白在内质网蓄积[5]。本例患者尿酸高且伴肾囊肿,需考虑ADTKD,但父亲仅有肾囊肿而无肾功能不全,不符合该病典型表现,确诊需基因检测。(4)髓质海绵肾:主要表现为髓质区集合管囊性扩张,可伴发血尿、感染、梗阻、钙质沉着症和尿路结石,多数为非遗传性,仅5%病例为家族性,该例患者临床无以上症状,因此不符合该种疾病。(5)肾小球囊肿病:顾名思义,其突出表现为肾小球囊肿。它包括一类疾病,其中由遗传因素导致的主要是HNF-1β基因突变,这与该例患者的肾脏病理表现和基因检测结果亦不符。此外,还有一些相对少见的类型,比如色素血管性斑痣性错构瘤病、结节性硬化等。这些疾病的鉴别除了病史和临床表现的区别,主要依赖于基因检测。该例患者经二代基因测序,结合其家族史,最终考虑为NPHP1基因突变相关的NPHP。
NPHP于1945年首次被报道[6],发病率在1/50 000-1/900 000之间,不同国家和地区之间存在差异;由于基因检测的普及率不同,该发病率可能有所低估。NPHP没有性别差异,分别占欧洲和北美儿童ESRD总数的10%~25%和5%左右。NPHP是一种常染色体隐性遗传性肾病,30%~40%的患者能鉴定出致病基因。目前已鉴定出26种基因的异常可导致该种疾病,由于这些基因编码的蛋白与纤毛生成或纤毛功能相关,因此又称为纤毛病[7]。有学者认为NPHP患者由于胞内信号通路缺陷导致纤毛功能受损,纤毛力学感受器异常使肾小管上皮细胞不能感知管腔内液体的流速,导致液体调控失调,出现肾囊肿。此外,NPHP1,INVS和NPHP4基因编码的蛋白也位于细胞连接处,能调节细胞间紧密连接。NPHP1基因是首个发现的导致少年型NPHP的基因,位于常染色体2q13[7],包含20个外显子,编码肾囊素蛋白(nephrocystin,也称nephrocystin-1)。NPHP1基因突变也是最常见的导致NPHP的基因突变类型,占20%~25%,其中又以大片段纯合缺失突变为主[9-10]。
NPHP根据临床发病年龄的不同可分为婴儿型,少年型和青年/成人型,其分型依赖临床表现和肾脏超声。婴儿型在胎儿时期即可出现母亲羊水过少,婴儿四肢挛缩、肺动脉发育不全和面部畸形,3岁前即进展至ESRD;肾脏超声表现为肾脏体积大伴肾囊肿,皮质回声增强。少年型最常见,表现为多饮多尿、遗尿、生长发育迟缓、慢性贫血,以及慢性肾脏病,通常不伴高血压,进展至ESRD的中位年龄是13岁;肾脏超声示正常或偏小的肾脏,皮髓界限不清、回声增强,皮髓交界囊肿形成。青年/成人型与少年型临床表现及超声检查结果类似,进展至ESRD的中位年龄是19岁[11]。此外,NPHP基因突变具有表型异质性,即10%~20%的NPHP患者可出现其他系统受累的表现,如视网膜色素变性、肝纤维化、骨骼异常和大脑发育异常,表现为各种临床综合征,如Joubert综合征,Senior-Løken综合征,先天性眼球运动障碍(COMA),COACH(小脑蚓部发育不良导致小脑性共济失调、脑膨出、眼球内组织缺损、肝纤维化、肾囊肿)等等。虽然这些临床综合征可归因于单个基因,但单个NPHP基因突变确实可以导致不同的临床表型。导致临床表型差异的原因尚不清楚,可能是由于基因突变的具体位点差异所导致。同时,在某些患者中还能见到寡基因、双基因以及三等位基因遗传的特点,这也能解释临床表型的异质性。NPHP1基因突变患者确诊的主要原因是多尿或者发现慢性肾脏病。这些患者的肾功能多在8~16岁迅速恶化,进展至ESRD的平均年龄是11.4岁。与其他基因突变相关的肾消耗病比起来,NPHP1基因突变患者的肾囊肿发生的年龄更大,平均年龄是12.3岁,肾外受累更少见[12]。
NPHP的肾脏病理表现并不特异,主要表现为肾小管基膜不规则增厚或变薄、分层,肾小管萎缩,间质纤维化、炎细胞浸润,部分病例可见肾小管囊样扩张[13]。肾活检虽然并非诊断所必须,但对有些病例仍然是非常有必要的,因为多数病例并不能发现致病基因。如果临床高度怀疑NPHP,但基因检测结果为阴性或无法开展基因检测,或临床诊断不明确,需鉴别是否存在其他肾脏疾病,均需行肾活检。值得注意的是,尽管NPHP本身不会导致高血压,但是肾功能衰竭后会出现高血压,对既往经肾活检确诊为高血压肾损害的年轻肾衰竭患者行基因检测,发现部分病例为纤毛病相关的基因突变[14]。针对NPHP,目前主要是支持治疗,重点是关注水电解质平衡、纠正贫血,以及控制高血压。
小结:NPHP作为一种遗传病,更多见于儿童,其诊断主要依赖于临床表现、实验室检查和肾脏超声检查,肾脏病理检查则对于鉴别排除其他肾脏疾病也有一定的诊断意义。由于近2/3病例目前并未发现致病基因,二代测序基因检测只能作为一种辅助确诊手段。对NPHP致病的遗传学基础和发病机制还有待进一步研究。
