体细胞突变在自身炎症及免疫性疾病发生中的作用
2022-11-22综述刘志红审校
龚 丞 综述 刘志红,2 审校
根据来源基因突变主要分为生殖系突变与体细胞突变两种。生殖系突变通过遗传获得,随着个体发育传递给所有细胞,其引发的疾病具有早发性、全身性的特点,并符合家系遗传模式。体细胞突变发生在受精卵形成后及个体生命过程中任意时刻,造成单个细胞的突变并传递给随之扩增的单克隆细胞群,通常不遗传给后代,由体细胞突变引发的疾病发病相对较晚且具有较高的异质性[1-2]。1882年,夜间阵发性血红蛋白尿(PNH)被定义为一种新的疾病,随后从部分PNH患者的造血干细胞中鉴定出了PIGA基因突变,PIGA基因编码的蛋白促进糖基磷脂酰肌醇向细胞膜转运,该基因突变导致补体异常激活并发生溶血,这也是首次在人体内证实体细胞突变可以引发疾病[3-4]。既往研究认为体细胞突变使细胞发生恶变倾向,主要与恶性肿瘤的发生发展密切相关[5]。随着测序技术的发展,研究者们发现许多非恶性增殖性疾病同样由体细胞突变引起[1,6],其中免疫调节异常因表现复杂、机制多样而受到特别关注。本文就体细胞突变引发自身炎症及免疫性疾病的研究进展做一综述。
体细胞突变的发生与调控
受到各种内源或外源性因素的影响,遗传物质可以发生多种变异,包括单碱基突变、短片断插入缺失突变、拷贝数变异、转座重组、染色体结构变异及非整倍型改变等,其中以单碱基突变为最常见的形式[7]。人类基因组共有约30亿个碱基对,近年研究发现,在人体内突变发生的频率约为每个细胞分裂每个碱基对1~5×10-10次[8-9],考虑到人类基因组庞大的碱基数量及持续存在的细胞分裂活动,突变在体细胞中的产生与累积是普遍存在、时刻发生的。
体细胞突变的机制主要包括DNA复制错误、DNA修复机制异常、直接DNA损伤等。DNA复制错误是复制过程中随机发生的异常事件,包括碱基错配、染色体错位、染色质断裂及不规则链合成等(图1)[10]。当DNA复制错误发生时,细胞将启动多种机制对其进行修复[11],而部分无法进行修复的复制错误,通常将引发细胞衰老与凋亡过程,从而阻止变异的累积。例如BRCA1基因编码的蛋白具有在细胞周期调节DNA修复的功能,其突变导致机体细胞发生变异增加[12],端粒酶基因TERT的突变与体细胞突变引起的克隆性造血存在关联[13]。直接DNA损伤可来源于多种刺激因素,DNA受到刺激发生单链或双链断裂、交联或碱基修饰,最终未能修复的损伤经由DNA复制过程产生体细胞突变[10-11]。
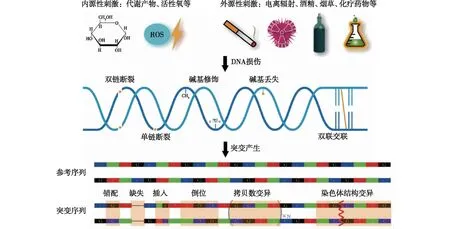
图1 体细胞突变发生的分子机制[10-11]
体细胞突变与疾病的关联及意义
由于体细胞突变发生的随机性,突变细胞的比例、类型、组织分布均存在个体差异,其临床表型更为多样。相较于生殖系致病突变,体细胞突变的表现可能局限于特定组织,缺乏疾病的某些典型特征,或是症状较轻甚至几乎正常。例如由体细胞突变引起的Alport综合征,患者常无症状或症状不典型, 为明确诊断带来困难[14-15]。在突变的检出方面,体细胞突变的分布及比例不能确定,采用收集血液、唾液或毛囊等较为易行的方式往往难以发现,侵入性检查的开展困难与对细胞分群与深度测序的依赖造成了体细胞突变检测的难点[16]。另外,特定基因的体细胞突变已成为肿瘤治疗研究热点。如BRCA1/2基因突变与特定种类恶性肿瘤的发生相关,携带BRCA1/2突变的肿瘤细胞缺乏DNA双链断裂修复机制,并展现出对铂类耐药等特性。2020年,美国临床肿瘤协会(ASCO)在指南中明确提出所有卵巢上皮肿瘤患者应当接受BRCA1/2及其他易感基因检测,对于未检出生殖系突变的,应当接受肿瘤BRCA1/2体细胞突变检测[17]。由此可见,体细胞突变不仅与疾病表现的异质性有关,还将对治疗反应造成影响。
体细胞突变与自身炎症性疾病
自身炎症性疾病是一种主要由机体固有免疫系统失调导致的疾病。不同于自身免疫性疾病,自身炎症以周期性发热及系统性炎症为特征,主要累及髓系细胞,同时缺乏自身免疫病的多种典型表现,如自身抗体产生、淋巴细胞异常激活等[18]。单基因自身炎症病被认为是具有明确致病突变的疾病类型,炎症通路相关基因发生突变,使相应信号分子的功能丧失或异常激活,进而直接引发疾病表型。而随着基因组测序技术的发展,越来越多的病例提示仅在部分体细胞中存在的有害突变同样可以引发类似表型,单基因自身炎症病的病因也不再仅限于生殖系突变。
炎症小体病是由炎症小体相关基因发生功能获得性突变引发的自身炎症性疾病,已报道的致病基因包括NLRP3、NLRP1、NLRC4、MEFV等。NLRP3是炎症小体的一部分,作为感受器响应病原体入侵,激活caspase-1及其下游信号分子。NLRP3功能获得性突变引发的疾病表型多样,统称为cryopyrin相关周期性综合征(CAPS)或新生儿起病的多系统炎症性疾病(NOMID),此类疾病主要通过显性遗传模式或新发突变致病。2015年,Nakagawa等[19]招募了56例具有CAPS或类似临床表现,但NLRP3基因测序结果显示未携带已知生殖系致病突变的患者,通过二代测序进一步分析发现其中7例患者(12.5%)存在6个不同的体细胞突变,均为错义突变且所占比例不等(5.5%~34.9%)。体外实验证明新发现的突变相较野生型均能显著激活核因子κB(NF-κB)信号通路并增强人单核细胞株THP-1的细胞死亡。在具有CAPS临床表现的患者中,可能有相当比例的体细胞突变携带者未能获得遗传诊断,提示深度测序的重要性。NLRC4同样具有识别病原体特定蛋白的功能,激活的NLRC4组成炎症小体并介导半胱氨酸天冬氨酸蛋白酶1 (caspase-1)活化,携带特定NLRC4生殖系突变的患者存在炎症小体的过度激活,并以自身炎症为主要表现[20]。2022年,Wang等[21]首次发现并验证了低比例的NLRC4体细胞突变可引起晚发性自身炎症性疾病。对患者的外周血DNA样本进行测序检测,发现其携带有很低比例的NLRC4(p.His443Gln)突变,对患者外周血单个核细胞(PBMCs)进行数字微滴PCR(ddPCR),结果显示单核细胞具有最高的突变比例(5.69%),中性粒细胞(2.10%)、B细胞(2.08%)、T细胞(0.66%)则相对较低,而在患者毛囊组织与正常人PBMCs中均未发现该突变。H443Q突变破坏了NLRC4自抑制结构,引起持续的炎症小体激活,患者血清白细胞介素1(IL-1)、IL-18及部分炎症因子水平较正常人升高,PBMCs中炎症相关基因表达上调,单细胞转录组测序结果显示在单核细胞中炎症激活相关信号通路存在特别富集,进一步提示该突变与疾病的关联。不同于生殖系突变,在体细胞突变的分析中,除了检出低频突变的技术手段,临床样本的合理收集也尤为重要。对于血液细胞突变,在常规采集血液的基础上,有必要进行明确的细胞分群,而对于怀疑其他组织的体细胞突变,则应考虑相应部位的采样。
自身炎症性疾病一般在新生儿期即迅速起病,而成人起病表现则更为多样,其中仍有多数患者缺乏明确的遗传诊断。2020年的一项研究在表型各异的成人起病的自身炎症性疾病患者队列中,以“基因型主导”的研究策略,通过对外周血进行二代测序分析,发现其中25例携带UBA1基因41位甲硫氨酸体细胞突变。UBA1基因位于X染色体,编码一种主要的E1酶,具有启动泛素化修饰的作用。此前研究证明异常泛素修饰与炎症性疾病发病存在密切联系,目前已发现数个相关基因,其功能异常可引发自身炎症表型。携带UBA1体细胞突变的患者出现成人起病、严重、难治性的全身炎症性疾病,主要表现为发热、血细胞减少、骨髓异常增殖、嗜中性皮肤病、肺部炎症、软骨炎、血管炎等,骨髓染色可见髓系细胞内典型空泡形成。对患者各类群细胞进行测序分析,发现UBA1突变主要见于外周血髓系细胞,而在淋巴细胞少见,提示特定细胞类型与炎症性疾病发生的关联。进一步通过在细胞系内过表达UBA1,证实41位甲硫氨酸突变可以导致UBA1一种亚型不能正常表达,该亚型主要定位于胞质,从41位甲硫氨酸起始翻译过程。突变使该亚型缺失起始密码子,产生从67位甲硫氨酸起始翻译的截短蛋白,该蛋白不具有正常的酶活性,携带突变的细胞泛素化过程受到抑制,从而激活免疫相关通路。另外,在斑马鱼模型中敲除UBA1胞质亚型将激活系统性炎症反应,证实UBA1功能异常在炎症性疾病发生中的作用。根据致病突变的遗传特征以及疾病表型特点,研究者将由UBA1体细胞突变引发的这一类自身炎症性疾病命名为液泡性、E1酶相关、X染色体连锁、自身炎症性、体细胞突变(VEXAS)综合征[22]。
体细胞突变与自身免疫性疾病
自身免疫性疾病的特征是针对自身抗原产生病理反应,导致炎症、细胞损伤或功能紊乱,并引起相应临床表现。自身免疫性疾病常涉及多种细胞类型,发生于局部或累及全身多器官[23]。
自身免疫性淋巴细胞增生综合征(ALPS)是一种Fas相关淋巴细胞凋亡途径异常导致的自身免疫性疾病,主要临床表现包括非恶性淋巴组织增生、肝脾肿大、免疫缺陷、血小板减少等,ALPS常见的致病基因包括FAS、FASLG、CASP10。2004年,Holzelova等[24]首次发现并报道了发生于淋巴细胞中的体细胞FAS杂合突变可引发ALPS,携带突变的淋巴细胞不再受到Fas相关凋亡信号的调控,而Fas非依赖的调控机制不受影响,CD4-CD8-双阴性T细胞出现显著扩增,患者表型与既往由生殖系突变引发的ALPS类似。但体外淋巴细胞凋亡试验显示,相比于携带生殖系突变的ALPS患者,携带FAS体细胞突变的患者对凋亡刺激响应率更高。
RAS家族基因突变在恶性肿瘤中常见,而在2011年,Niemela等[25]与Takagi等[26]分别鉴定了数例携带KRAS基因G13D体细胞突变的患者,发现其主要表现为非恶性的白细胞功能异常与自身免疫。激活的KRAS突变可能通过抑制凋亡相关蛋白BCL-2作用使T细胞凋亡减弱,从而引发自身免疫。因其机制与ALPS类似但临床表现不同,作者提出将该种由RAS基因体细胞激活突变引起的疾病命名为RAS相关的类ALPS病(RALD)。2016年,Takagi等[27]报道了1例携带KRAS基因G13C突变的男性患儿,临床表现合并免疫性血小板减少、过敏性紫癜以及白塞氏病。突变主要存在于T淋巴细胞、B淋巴细胞及NK细胞中,而未见于粒细胞,该患者表型与此前报道的RALD存在差异,提示携带突变的细胞类型与疾病表型可能存在关联。
大颗粒淋巴细胞(LGL)白血病是一种淋巴增生性疾病,主要特征为CD3+CD8+细胞毒性T细胞(CTLs)异常增多、骨髓LGL浸润、中性粒细胞减少、常伴发自身免疫性疾病。LGL白血病本质上并不是一种恶性肿瘤,由自身免疫性T淋巴细胞攻击自身血液细胞导致血细胞减少。STAT3和STAT5B是LGL白血病的常见致病基因,2012的一项研究对76例患者的CTLs进行全外显子测序,发现其中31例(40%)携带STAT3体细胞突变,且突变均发生于基因21号外显子。结构模拟提示这些位点的氨基酸替换影响STAT3与DNA的结合力,体外实验同样证明突变的STAT3可导致受其调控的基因发生转录增强,提示突变可能通过激活CTLs引发疾病表型[28]。
慢性肉芽肿病(CGD)是一种以反复发生危及生命的细菌及真菌感染和肉芽肿形成为主要特征的免疫异常,其中约5%的患者合并系统性红斑狼疮、免疫性血小板减少症、幼年特发性关节炎等自身免疫性疾病。多数CGD患者携带CYBB基因突变,该基因位于X染色体,编码一种NADPH氧化酶亚基,CYBB表达异常导致NADPH氧化酶功能丧失,影响吞噬细胞抵抗病原菌能力,进而引发免疫异常表型。2005年,Wolach等[29]报道了1例女性晚发性CGD患者,外周血细胞测序结果显示患者携带CYBB(p.Tyr30_Arg31delinsTer)突变。检测mRNA发现绝大多数细胞仅表达突变的mRNA,提示另一条正常的X染色体发生了随机失活,后续实验也证实仅0.4%~2%的中性粒细胞具有正常的NADPH氧化酶活性。另外,对患者颊部细胞及记忆T细胞取样测序未检出该突变,提示该CYBB基因突变为粒细胞特异性的体细胞突变。
2017年一项关于类风湿性关节炎(RA)的研究发现患者具有较高的CD8+T细胞单克隆扩增水平,其中约有20%的CD8+T细胞存在免疫相关基因体细胞突变,而在CD4+T细胞则未检出同类突变。部分发生突变的基因,如SLAMF6、IRF1已被证实与自身免疫相关,提示免疫相关基因的体细胞突变在CD8+T细胞中的累积可能与RA的发生发展存在关联[30]。
Tn综合征是由于部分血细胞表面携带未完全糖基化的Tn抗原,从而引发的一种自身免疫性疾病。2005年的一项研究发现C1GALT1C1基因发生体细胞突变是Tn综合征发生的重要原因,其编码的Cosmc蛋白功能缺失将导致Tn抗原的异常糖基化,并产生自身免疫[31]。在IgA肾病中,IgA1分子表面的异常糖基化被认为是其分子机制之一,而Cosmc蛋白则行使IgA1分子半乳糖基化的功能,该基因突变与IgA肾病的发生可能存在关联。
体细胞突变的检测
在临床工作中,不同于生殖系突变导致的遗传性疾病,体细胞突变缺乏明确的家系遗传模式,其导致的疾病常无家族史可循,为遗传诊断带来一定困难。近年来,随着二代测序技术的发展与普及,全外显子组测序(whole exome sequencing,WES)、全基因组测序(whole genome sequencing,WGS)、靶区域测序(target sequencing)、单细胞全基因组测序等技术的应用大大推动了体细胞突变相关研究。而在实际工作中,对于可疑患有遗传性疾病的患者组织样本,WES与WGS以其简便的样本采集,成熟的分析流程与较低的价格成为目前遗传诊断主流的技术方法。体细胞突变的检测流程主要包括(1)测序与比对。测序得到的原始序列经过质控,将可信的序列片段匹配到已知正常人类基因组序列的对应位置,在匹配序列中可能包含部分位置的错配。(2)突变检测。匹配到基因组的序列片段经过去重复,得到基因组各区域的覆盖深度,同时借助基因或功能区域在基因组中的已知位置完成突变的识别。对于生殖系杂合突变,正常序列与突变序列比例大约为1∶1,而对于体细胞突变,突变序列的比例往往显著低于正常序列,甚至在一些覆盖深度较低的区域,体细胞突变可能无法检出。另外,由于需要鉴别由测序质量不佳带来的假阳性,有必要在后续分析中应用一代测序、PCR等低通量手段或深度更高的靶区域测序进行重复验证。(3)突变注释与解读。检测得到的突变条目仅包含本身位置与质量信息,为了解突变的性质,需要借助多个数据库,对该突变的致病性软件评分、人群频率、既往研究情况等各方面进行注释。在突变解读方面,除临床医生的个人判断外,美国医学遗传学与基因组学学会(ACMG)制订了关于孟德尔遗传病变异解读标准与指南,对突变通过多种证据进行综合评价,得到可供参考的突变致病性分级[32]。
小结:体细胞突变可能发生于个体生命周期的各个阶段,并具有随机性,可发生于多种细胞,即使携带同一基因突变的患者,在发病时间、临床表现等方面也具有很大差异。与生殖系突变的直接对应关系不同,体细胞突变发生的时间、细胞微环境、突变比例均可能影响疾病的表型。对于体细胞突变的发现及致病机制研究有助于更好地从分子、细胞层面认识特定疾病。体细胞突变在自身炎症与自身免疫性疾病发病机制中起着关键性作用(表1)。对体细胞突变的研究极大地丰富了对基因变异与疾病之间的认识,对于疾病的精准诊断、分型、治疗具有重要意义。

表1 体细胞疾病引发的自身炎症及免疫性疾病
