超高效合相色谱法测定鱼肉中氟苯尼考对映体及其代谢产物残留量
2022-10-31张文华施雅梅叶芮辰徐敦明伊雄海
张文华,吴 媛,施雅梅,汪 鹏,叶芮辰,徐 可,谢 文,徐敦明,伊雄海
(1.杭州海关技术中心,浙江 杭州 310016;2.浙江省检验检疫科学技术研究院,浙江 杭州 310016;3.厦门海关技术中心,福建 厦门 361026;4.上海海关动植物与食品检验检疫技术中心,上海 200135)
氟苯尼考(florfenicol,FF)又称氟甲砜霉素,为甲砜霉素的单氟衍生物,是新一代氯霉素类动物专用广谱抗生素,其主要代谢产物为氟苯尼考胺(florfenicol amine,FFA),广泛用于防治水产养殖过程中的细菌性疾病。FF的残留包含原形药物及其主要代谢产物FFA。FF的化学结构中有2 个手性碳原子,即存在4 个对映体结构,而在实际生产中只能合成两个对映体(图1)。其中FF(1、2)左旋体构型具有抗菌活性,而FF(1、2)右旋体构型无活性甚至对FF的活性有一定的抑制作用。然而,GB 31650—2019《食品中兽药最大残留限量》仅规定了鱼肉中FF外消旋体的最高残留限量≤1.0 mg/kg,并未对具有抗菌活性的左旋FF和无活性的右旋FF的残留限量进行精确规定。如果能进一步精准测定FF对映体及其代谢物FFA在鱼肉中的残留量,将能为FF药物残留分析提供精确的判定。

图1 (-)-FF(A)、(+)-FF(B)和FFA(C)的结构式Fig.1 Chemical structures of (-)-FF(A),(+)-FF(B) and FFA (C)
目前国内外测定兽药的方法主要有高效液相色谱法、高效液相色谱-串联质谱法等方法;而检测FF对映体的方法主要为高效液相色谱法,该方法分离度好,但存在分析时间长,有机试剂消耗量大等问题。近年来新推出的超高效合相色谱技术(ultraperformance convergence chromatography,UPC)引起大家的广泛关注,该技术的主要流动相为超临界二氧化碳(CO),能精确调控分析物的分离度和保留时间,主要用于拆分和定量结构类似物、异构体等,尤其在分离手性化合物方面具有分离效率高、有机试剂消耗量少的独特优势,弥补了传统液相色谱系统在对映体分析功能上的不足。目前,尚未见到将UPC技术应用于FF对映体的拆分及残留测定的报道。
为解决传统液相色谱法拆分对映体存在的问题,本实验建立一种UPC的方法,用于拆分和测定FF对映体及其代谢物;对FF对映体及其代谢物FFA的仪器色谱分离条件和鱼肉中FF对映体的净化条件进行优化;在最优条件下,将建立的UPC方法应用于实际鱼肉样品和FF外消旋体标准品并进行分析。该方法具有分析速度快、分离效果好、有机溶剂消耗少等特点,以期为指导FF兽药的生产过程、质量控制和药效评价以及鱼类产品的质量安全监管提供一定技术支持。
1 材料与方法
1.1 材料与试剂
鱼肉购于当地超市和农贸市场,粉碎后于-18 ℃条件下保存。Anpel FF外消旋体标准品(质量浓度100 mg/L)上海安谱实验科技股份有限公司;BePure FF外消旋体标准品(纯度99.8%)、FFA标准品(纯度≥99.7%) 北京曼哈格生物科技有限公司;FF外消旋体标准品(纯度99.0%) 德国Dr.Ehrenstorfer公司;(-)-FF标准品、(+)-FF标准品(纯度≥99.0%) 中国牧工商(集团)总公司研究院。
乙腈、甲醇、甲酸、乙醇、异丙醇、正庚烷、乙酸乙酯(均为色谱纯) 西班牙萨劳有限公司;氨水浙江杭州高晶精细化工有限公司;磷酸氢二钾、磷酸二氢钾 广东汕头西陇科学股份有限公司;CO(纯度99.999%) 上海宝钢普莱克斯实用气体有限公司;除特殊说明外,所有试剂均为分析纯。
配制磷酸盐缓冲液:称取8 g KHPQ和2 g KHPO溶解混合,用超纯水定容至1 000 mL。
1.2 仪器与设备
Acquity UPC仪(带有PDA二极管阵列检测器)、Oasis MCX混合型阳离子交换固相萃取小柱(3 mL,60 mg) 美国Waters公司;CHIRALPAK AD-3手性色谱柱(150 mm×3.0 mm,3 µm) 大赛璐药物手性技术(上海)有限公司;AE260电子天平 梅特勒-托利多仪器(上海)有限公司;R215旋转蒸发仪 瑞士Büchi公司;ELGA CLXXXUVM2超纯水净化系统 英国ELGA LabWater公司;MS2涡旋混匀器 上海医大仪器厂;N-EVAP111氮吹仪 日本东京理化器械株式会社;Oasis HLB聚合物固相萃取小柱(3 mL,60 mg) 杭州金谱科学仪器有限公司;Supleco C固相萃取小柱(3 mL,300 mg) 上海安谱科学仪器有限公司。
1.3 方法
1.3.1 标准溶液的配制
分别准确称取0.01 g(精确至0.1 mg)3 份市售FF外消旋体标准品,用甲醇溶解并定容至10 mL,配制成1.0 g/L FF外消旋体标准储备液。分别准确吸取一定体积的标准储备液,用正庚烷-异丙醇(8∶2,/)溶液将其稀释成10.0 mg/L FF外消旋体标准中间溶液。
分别准确称取0.01 g(精确至0.1 mg)(-)-FF、(+)-FF和FFA标准品,用甲醇溶解并定容至10 mL,配制成质量浓度为1.0 g/L的(-)-FF、(+)-FF和FFA标准储备液。分别量取一定体积的标准储备液,用异丙醇稀释并定容至10 mL,配制成100.0 mg/L的混合标准溶液。1.0、2.0、4.0、10.0、20.0、40.0 mg/L标准工作溶液用正庚烷-异丙醇(8∶2,/)溶液逐级稀释混合标准溶液获得。
1.3.2 样品的制备
从全部鱼肉中取出有代表性的样品约500 g,用粉碎机粉碎,分别装入洁净容器,密封标记。将鱼肉样品于-18 ℃冷冻保存。
1.3.3 样品前处理
1.3.3.1 样品提取
称取10.0 g(精确至0.01 g)鱼肉样品置于50 mL具塞离心管中,加入25 mL 2%氨水-乙酸乙酯溶液(/),涡旋混匀1 min,以8 000 r/min离心5 min,吸取上清液至浓缩瓶中,以25 mL乙酸乙酯重复提取,合并两次提取液。在40 ℃以下水浴减压浓缩提取液至近干,加10 mL 1%甲酸-水溶液(/)分次溶解残渣,转移至50 mL具塞离心管中,加入15 mL正己烷,涡旋混匀。以8 000 r/min离心5 min,弃掉上层正己烷,重复向离心管中加入15 mL正己烷脱脂,弃掉上层正己烷,下层水相待净化。
1.3.3.2 样品净化
HLB、C固相萃取小柱使用前依次用3 mL甲醇和3 mL水活化;MCX固相萃取小柱使用前依次用3 mL甲醇和5 mL 1%甲酸-水溶液(/)活化。
移取1.3.3.1节待净化的下层水相于活化好的MCX固相萃取小柱中,用5.0 mL甲醇-水溶液(1∶1,/)淋洗,抽干后用6 mL 5%氨水-甲醇洗脱,收集洗脱液。将洗脱液于40 ℃以下水浴中氮气吹至近干,向离心管中加入1 mL正庚烷-异丙醇溶液(8∶2,/),涡旋溶解充分,过0.22 μm滤膜后装入进样小瓶中,待测。
1.3.4 UPC条件
CHIRALPAK AD-3手性色谱柱(150 mm×3.0 mm,3 µm);检测波长:224 nm;系统背压:13.8 MPa;柱温:40 ℃;流动相:A 为超临界CO,B 为氨水-甲醇溶液(0.5∶99.5,/);梯度洗脱程序:0~1.5 min,90% A、10% B;1.5~2.5 min,90%~77% A、10%~23% B;2.5~5.5 min,77% A、23% B;5.5~5.8 min,77%~90% A、23%~10%B;5.8~8.0 min,90% A、10% B;流速:1.0 mL/min;进样量:5.0 µL。
1.3.5 标准溶液稳定性测定
对新配制FF对映体及其代谢物FFA标准溶液进行考察:与分别贮存不同时间后的FF对映体和FFA的含量比较,观察FF对映体和FFA标准溶液的稳定性。分别准确移取1.0 mL 10.0 mg/L FF对映体和FFA的混合工作溶液于7 个带划痕的1.5 mL UPC专用进样小瓶中,上机进行含量测定,检测后再转移至7 个带铝盖密封的进样小瓶中,并用封口膜封好后置于-18 ℃保存。按照优化后的色谱条件测定新配制的与分别贮存1、3、5、7、14、30、60 d的FF对映体和FFA标准溶液。
1.3.6 加标回收率及精密度测定
分别向不含有FF和FFA的鱼肉空白样品中添加标准溶液进行添加回收率和精密度测定。(-)-FF和(+)-FF的添加量分别为0.1、0.2、1.0 mg/kg,FFA的添加量分别为0.2、0.4、1.0 mg/kg,平行测定6 次,计算加标回收率及相对标准偏差(relative standard deviation,RSD)。
1.4 数据处理
采用SPSS 12.0软件处理数据,用检验评价新配制与贮存不同时间的FF对映体和FFA标准溶液含量的差异显著性。
2 结果与分析
2.1 UPC2条件的优化
2.1.1 流动相中助溶剂的优化
UPC分析中,通常使用超临界CO为主要流动相,使用少量有机溶剂作为助溶剂,以调节对目标产物的选择性和洗脱能力。在最佳条件下,考察氨水-甲醇溶液(0.5∶99.5,/)、甲酸-甲醇溶液(0.5∶99.5,/)、甲醇3 种助溶剂对10 mg/L标准溶液中两种FF对映体和FFA分离的影响。由图2可知,当使用甲酸-甲醇溶液时,基线升高;当使用甲醇时,两种FF对映体实现了基线分离,但FFA未能出峰;当选择氨水-甲醇溶液作为助溶剂时,两种FF对映体和FFA在5.0 min内实现完全分离。这是因为FFA含有氨基,属于极性较强的化合物,而氨水的加入能够延长出峰,明显改善FFA的响应和峰形。因此,本实验选择氨水-甲醇溶液(0.5∶99.5,/)作为助溶剂。
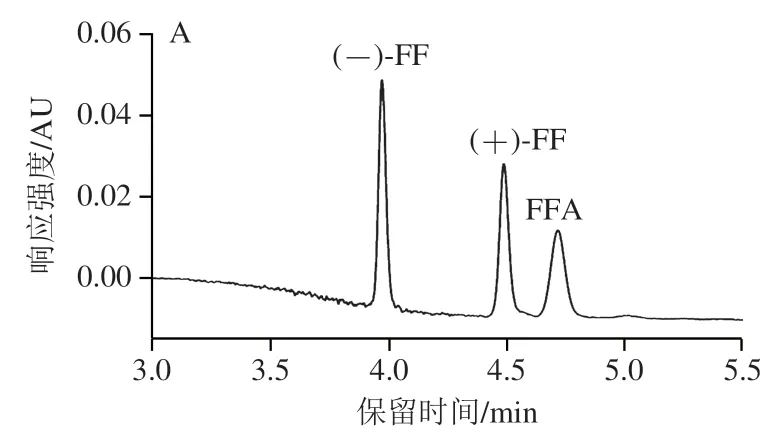
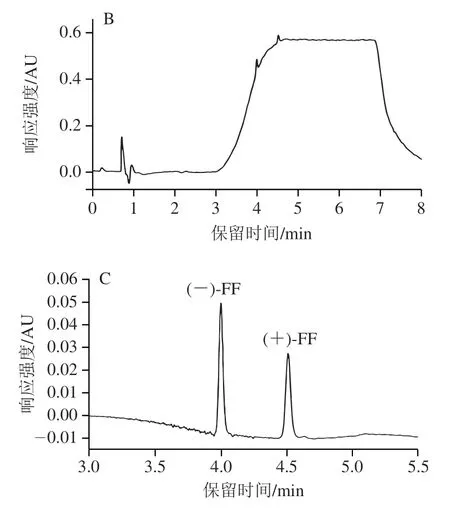
图2 不同助溶剂对(+)-FF、(-)-FF和FFA分离效果的影响Fig.2 Effect of different cosolvents on the separation of (+)-FF,(-)-FF and FFA
2.1.2 系统背压的优化
通过柱温和系统背压的调节可以改变UPC系统中超临界CO的密度,从而改变流动相对物质的洗脱能力、溶解能力和选择性。CO的温度超过31 ℃且压力超过7.38 MPa时,CO才会进入超临界状态。因此,在最佳条件下,考察系统背压在10.3~24.1 MPa范围内对10 mg/L标准溶液FF对映体和FFA分离的影响。如图3所示,随着系统背压的降低,3 个目标物的保留时间延长;3 个目标化合物在背压13.8 MPa时的色谱峰形和分离度均达到最佳。综合考虑保留时间、系统压力及峰形,本实验选择系统背压为13.8 MPa。
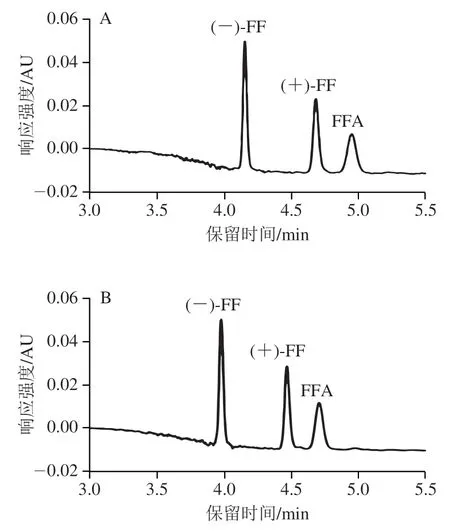
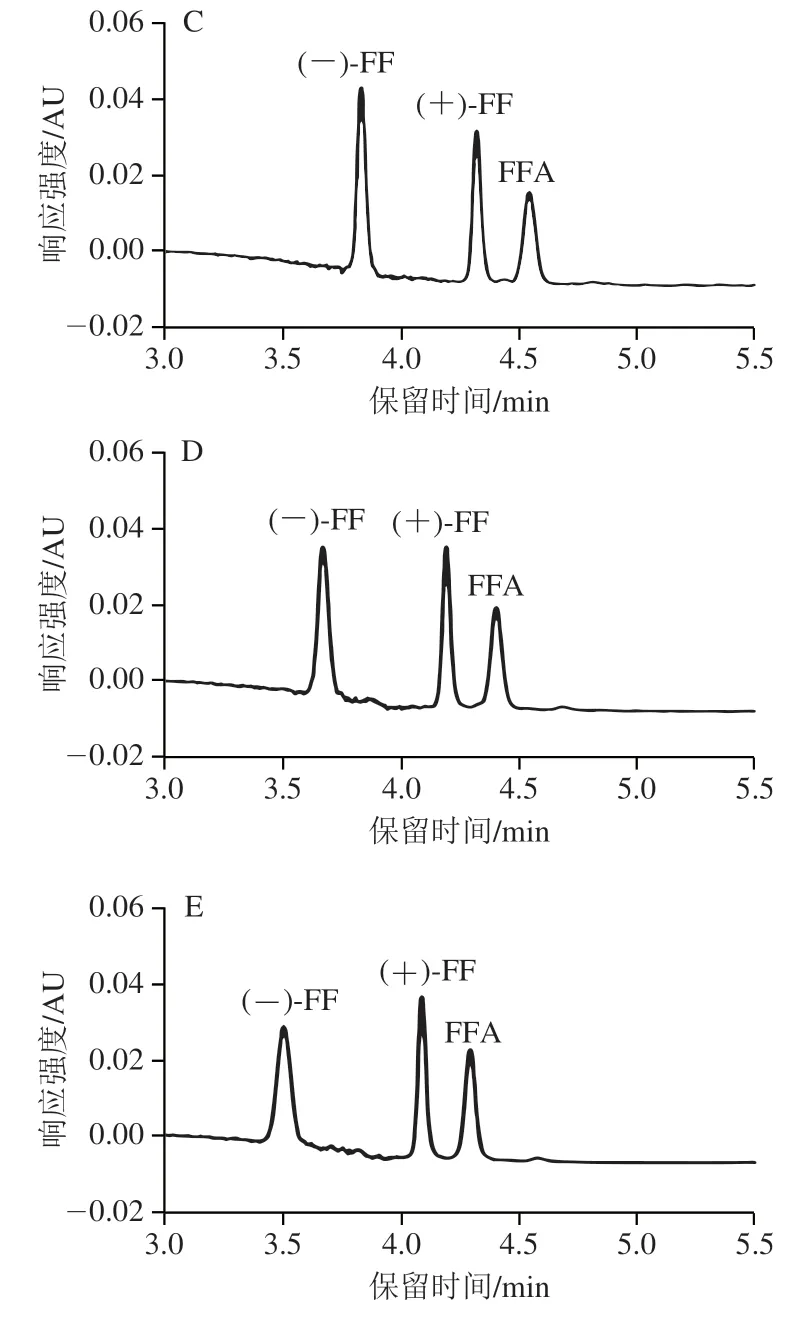
图3 不同系统背压对(+)-FF、(-)-FF和FFA对映体分离效果的影响Fig.3 Effect of the system’s back pressure on the separation of(+)-FF,(-)-FF and FFA
2.1.3 色谱柱温度的优化
一般通过调节UPC系统中色谱柱温度改变流动相密度,从而影响对目标物的分离效果。随着色谱柱温度降低,超临界CO流体的密度增大、黏度增高,对目标物的洗脱能力也随之增大,保留时间缩短。考虑到CHIRALPAK AD-3手性色谱柱的最高推荐运行温度(40 ℃)和超临界CO的条件(压力>7.38 MPa,温度>31 ℃),在最佳条件下,考察色谱柱温度在31~40 ℃范围内对10 mg/L标准溶液中两种FF对映体和FFA分离的影响。结果表明,随着柱温降低,目标物的保留时间逐渐缩短;当柱温为31 ℃和35 ℃时,(+)-FF和FFA的分离度不佳,部分色谱峰出现重叠,分离度分别为0.81和0.83;继续升高柱温至40 ℃时,(+)-FF和FFA的分离度为1.8,3 个目标化合物在5.0 min内实现良好的基线分离。因此,本实验选择40 ℃作为色谱柱温度。
2.2 鱼肉样品前处理的优化
2.2.1 提取试剂的优化
通常采用氨化乙酸乙酯、乙酸乙酯、水-丙酮溶液(2∶8,/)、乙腈、甲醇对样品中FF进行提取。参照1.3.3节的前处理方法,使用这4 种试剂提取鱼肉样品中的FF及FFA,经MCX固相萃取小柱净化后,上机检测,考察其提取效果。结果表明,采用水-丙酮溶液(2∶8,/)、乙腈和甲醇提取时,FFA均未出峰,干扰峰较多;采用乙酸乙酯提取时,FFA的回收率较低,存在干扰峰;在乙酸乙酯中加入氨水能够明显提高FFA的回收率,且干扰峰较少,与文献[34]相符,这是因为FFA含有氨基且极性较强。因此,本实验采用氨化乙酸乙酯作为提取试剂。
2.2.2 净化条件的优化
经最优试剂提取后,使用C、HLB、MCX3 种常用固相萃取小柱分别对待净化的鱼肉样品的下层水相进行净化(C、HLB柱采用1.1节中磷酸盐缓冲溶液溶解上柱,水淋洗,甲醇洗脱),上机检测,考察其净化效果。结果表明,MCX固相萃取小柱的净化效果最好,且(-)-FF、(+)-FF和FFA回收率依次为90.0%、98.2%和94.1%;HLB固相萃取小柱的回收率略优于MCX固相萃取小柱,但是经MCX固相萃取小柱净化得到的色谱图中干扰峰较少,与文献[28,41]相符。综合考虑,本实验采用MCX固相萃取小柱对鱼肉样品中FF对映体和FFA进行净化。
2.3 标准溶液的稳定性分析
为保证分析结果的准确度,对新配制与不同贮存时间后的FF对映体及其代谢物FFA标准溶液进行考察。结果表明:贮存7 d内(-)-FF、(+)-FF和FFA含量的RSD分别为3.3%、1.5%和4.2%;贮存14 d内(-)-FF、(+)-FF和FFA含量的RSD分别为3.5%、1.9%和10.2%;贮存30 d内(-)-FF、(+)-FF和FFA含量的RSD分别为10.8%、10.4%和15.2%。采用检验评价新配制与贮存不同时间的FF对映体和FFA含量的差异显著性。结果表明,与新配制的FF对映体标准溶液相比,在-18 ℃下贮存14 d的标准溶液中两种FF对映体含量差异不显著(>0.05),具有良好的稳定性;贮存30 d的标准溶液中两种FF对映体含量差异显著(<0.05),表现出较差的稳定性。与新配制的FFA标准溶液相比,在-18 ℃下贮存7 d的标准溶液中FFA含量差异不显著(>0.05),具有良好的稳定性;贮存14 d的标准溶液中FA含量差异显著(<0.05),表现出较差的稳定性。因此,两种FF对映体标准溶液在-18 ℃条件下可贮存14 d,FFA标准溶液在-18 ℃条件下可贮存7 d。
2.4 方法学验证
2.4.1 方法的线性范围和定量限(limit of quantitation,LOQ)
对(-)-FF、(+)-FF和FFA的系列混合标准溶液进行测定。以质量浓度为横坐标(),对应标准品的峰面积为纵坐标(),绘制标准曲线,求得回归方程和相关系数()。由表1可知,两种FF对映体在1.0~20.0 mg/L质量浓度范围内和FFA在2.0~40.0 mg/L质量浓度范围内均呈良好的线性关系,均大于0.999 3。通过在不含FF和FFA的鱼肉空白样品中添加标准品并进行测定,得到LOQ(信噪比10)。由表1可知,(-)-FF和(+)-FF的LOQ均为0.1 mg/kg,FFA的LOQ为0.2 mg/kg。

表1 各目标化合物的线性范围、线性方程、R2和LOQTable 1 Linear range,linear equation,R2 and LOQ of each target compound
2.4.2 加标回收率以及精密度分析
不加空白样品基质,在2%氨水-乙酸乙酯溶液中加入(-)-FF、(+)-FF和FFA 3 种标准品,经过1.3.3节的前处理后上机检测。结果显示,(-)-FF、(+)-FF和FFA的回收率依次为96.7%、98.1%、94.0%,RSD依次为5.2%、6.3%、6.8%,说明该方法具备良好的稳定性。
对添加回收鱼肉样品进行测定,计算加标回收率及RSD。由表2可知,3 种目标化合物的回收率范围为81.5%~108.0%,RSD范围为5.3%~9.3%。该回收率和精密度符合GB/T 27404—2008《食品理化检测》的要求,能够满足鱼肉样品的分析要求,可用于日常检测。

表2 鱼肉样品中FF对映体和FFA的加标回收率和RSD(n=6)Table 2 Spiked recoveries and RSDs of florfenicol enantiomers and florfenicol amine in fish meat (n=6)
2.5 建立的UPC2方法的应用
2.5.1 实际样品分析
为了考察方法的实用性和有效性,在最优条件下,应用所建立的方法对随机抽取的30 份市售鱼肉样品中FF对映体和FFA进行检测。结果表明:30 份鱼肉样品中均未检出(+)-FF;其中1 份鳊鱼样品中检出170 µg/kg (-)-FF;1 份黑鱼(乌鳢)样品中检出537 µg/kg FFA。
2.5.2 市售标准品的纯度分析
在最优条件下,应用所建立的方法对3 份市售常见FF外消旋标准品进行拆分及测定,结果如图4所示。3 份FF外消旋标准品中均未检出(+)-FF,Anpel、Bepure和Dr.Ehrenstorfer FF外消旋标准品中(-)-FF质量浓度分别为10.0、9.2、9.4 mg/L。市售Bepure和Dr.Ehrenstorfer两种外消旋固体标准品的纯度和实际测量值存在偏差,偏差值分别为8.0%和6.0%。
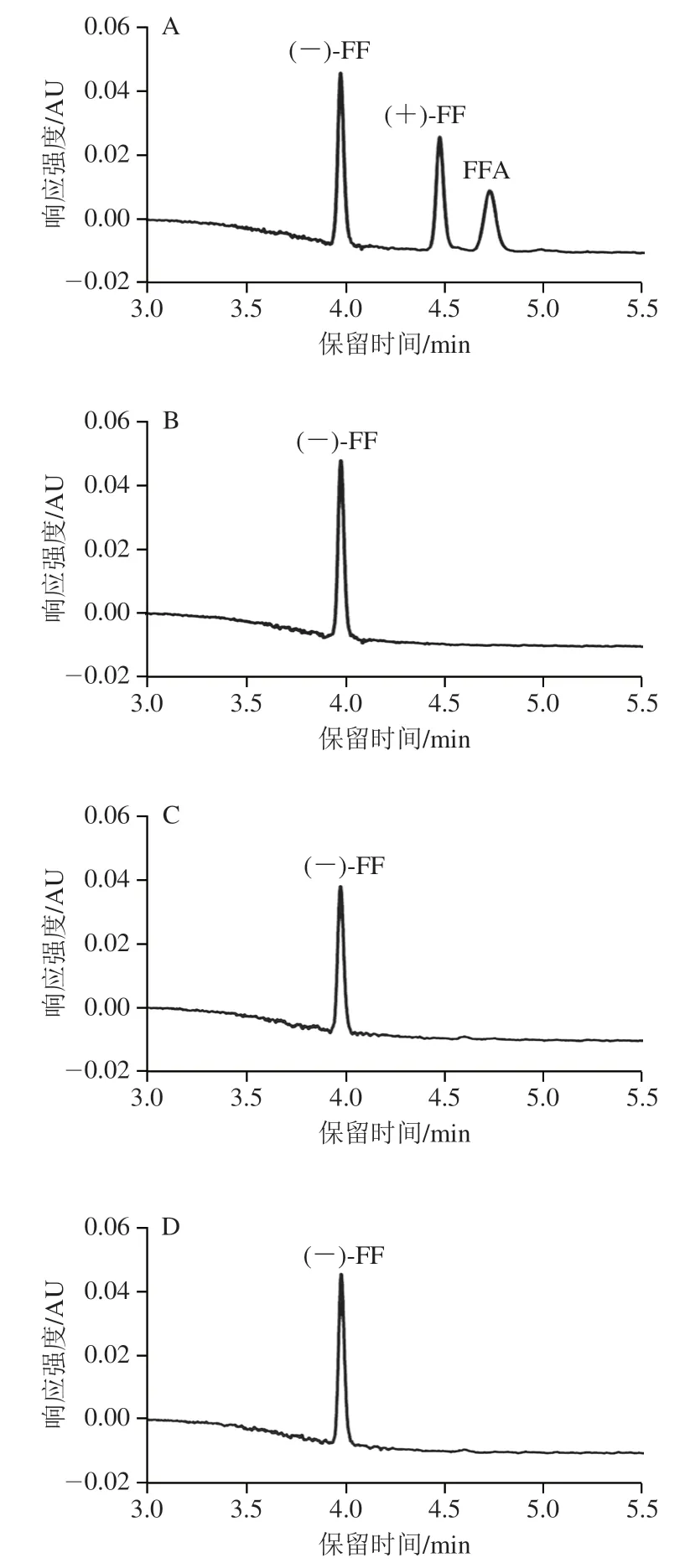
图4 FF对映体、FFA及外消旋体标准品的色谱图Fig.4 Chromatograms of florfenicol enantiomers,florfenicol amine and racemic florfenicol standard
市售常用FF外消旋体标准品和鱼肉样品的检测结果显示,均未检出(+)-FF,只检出(-)-FF和FFA,检测结果与文献[6,18,20]相符,故阳性鱼肉中代谢物质FFA是由(-)-FF对映体代谢产生。
3 结论
建立一种基于UPC的检测方法用于分离鱼肉样品中两种FF对映体和FFA。经分析得到此方法的最优条件:使用氨化乙酸乙酯对样品进行提取,通过MCX固相萃取小柱对样品进行净化后,采用CHIRALPAK AD-3手性色谱柱分离,以超临界CO(流动相A)和氨水-甲醇溶液(0.5∶99.5,/)(流动相B)为流动相进行梯度洗脱,系统背压13.8 MPa,柱温40 ℃。将最优条件用于后续实验。在0.1~1.0 mg/kg范围内进行加标回收实验,两种FF对映体和FFA的加标回收率为81.5%~108.0%,RSD为5.3%~9.3%。使用建立的方法对实际鱼肉样品和市售标准品进行了分析测定。结果表明,本方法具有分析速度快、准确度好、分离效果好等特点,能够满足鱼肉中左旋FF的纯度分析及快速定量需要。
