色谱-串联质谱法测定中药材中33种禁用农药残留量的方法探讨
2022-10-21刘俊俊罗秋红章亚玲高丽球万建春占春瑞
刘俊俊,蔡 丹,罗秋红,章亚玲,高丽球,韩 颖,万建春,占春瑞
(南昌海关技术中心,南昌 330038)
中药是我国宝贵的文化遗产,在医学领域发挥着越来越重要的作用。随着需求量的不断增长,人工种植中药材越来越多,随之而来的农药残留不仅危害人体健康[1-2],也是制约中药走向国际市场的瓶颈,因此加强中药材中农药残留的监测具有重要意义。《中华人民共和国药典》(2020版)(简称Ch P2020)四部通则中规定了33种禁用农药甲胺磷、甲基对硫磷、对硫磷、久效磷、磷胺、六六六、滴滴涕、杀虫脒、除草醚、艾氏剂、狄氏剂、苯线磷、地虫硫磷、硫线磷、蝇毒磷、治螟磷、特丁硫磷、氯磺隆、胺苯磺隆、甲磺隆、甲拌磷、甲基异柳磷、内吸磷、克百威、涕灭威、灭线磷、氯唑磷、水胺硫磷、硫丹、氟虫腈、三氯杀螨醇、硫环磷和甲基硫环磷,按照化学结构,其可被分为有机磷类、有机氯类和氨基甲酸酯类。有机磷类农药是一种含磷的广谱性杀虫剂,甲胺磷、甲拌磷、水胺硫磷等均为常见的有机磷类农药,具有不稳定、易分解、残留期短的性质,大多数有机磷类农药都有剧毒[3]。有机氯类农药是一种广谱性杀虫剂,常见的有六六六、滴滴涕,该类农药的特点是稳定性好、不易降解,且会通过生物链富集在动植物体内,危害人类健康及环境安全[4]。氨基甲酸酯类农药是在有机磷酸酯类之后发展起来的合成农药,是一类具有高效、易分解、残留期短特点的广谱性农药,应用于作物杀虫、杀螨、除草等,克百威、涕灭威是常用的氨基甲酸酯类农药,此类农药在空气和阳光下易分解,土壤中的半衰期较短,毒性较有机磷酸酯类低[5]。
Ch P2020四部通则第五法“药材及饮片(植物类)中禁用农药多残留测定法”中给出了直接提取法、Qu ECh ERS法和固相萃取法(分散型净化材料净化管、HLB柱和复合氨基柱)等3种样品前处理方法。直接提取法适用于基质相对简单的样品;QuEChERS法通用性高,但复杂基质共萃取物的净化效果不好;分散型净化材料净化管操作简单,但同样不适用于复杂基质样品的前处理;HLB柱可以去除部分叶绿素、挥发性油和磷脂,但对色素含量较高的物质净化效果不好;复合氨基柱对复杂基质样品的除杂效果较好,但对部分酸性农药可能有吸附作用。农药残留的检测方法有气相色谱法(GC)[6-7]、气相色谱-串联质谱法(GC-MS/MS)[8-10]、高效液相色谱-串联质谱法(HPLC-MS/MS)[11-12]等。其中,GC主要用于有机磷类和有机氯类农药的检测,但分离能力有限,不适用于多种农药的同时分析;GCMS/MS和HPLC-MS/MS具有较高的准确度、灵敏度和较好的抗干扰能力,可同时分析多种农药。针对不同中药材应采用哪种前处理方法、选用哪种检测技术,Ch P2020中未做阐述。
本工作采用GC-MS/MS和HPLC-MS/MS,测定了8种中药材中33种禁用农药的含量,对不同前处理方式、不同检测方法结果的差异性进行了探讨,以期为中药材农药残留检测技术的选择提供参考。
1 试验部分
1.1 仪器与试剂
U3000-TSQ Quantiva型液相色谱-三重四极杆质谱仪;7890-7000型气相色谱-三重四极杆串联质谱仪;IKA T25型均质器;R-215型旋转蒸发仪;BSA2202S型电子天平;ME155DU型电子天平;TDL-5-A型离心机;IKA MS3型涡旋振荡器;ELGA Q15型超纯水装置。
单标准储备溶液:分别称取适量的农药及其异构体或氧类似物标准品,根据各物质的溶解性分别采用甲醇、乙腈或丙酮溶解并定容至10 mL,配制成1.0 g·L-1单标准储备溶液。其中,三氯杀螨醇现用现配。
内标溶液:称取适量的磷酸三苯酯,用乙腈溶解并定容至10 mL,配制成100 mg·L-1内标溶液,应用于GC-MS/MS。
混合标准储备溶液:根据单标准储备溶液的定容液将其分为3组,分别移取适量的单标准储备溶液,用甲醇、乙腈或丙酮稀释并定容至10 mL,配制成10 mg·L-1混合标准储备溶液。
磷酸三苯酯、33种农药及其异构体或氧类似物标准品的纯度均大于98.0%;甲醇、乙腈为色谱纯;无水硫酸钠、无水硫酸镁、无水乙酸钠、甲苯均为分析纯;试验用水为超纯水。
1.2 仪器工作条件
1.2.1 HPLC-MS/MS
1)色谱条件 Agilent Eclipse Plus C18色谱柱(100 mm×2.1 mm,1.8μm);柱温35℃;流动相A为含0.1%(体积分数)甲酸的5 mmol·L-1甲酸铵溶液,B为乙腈;流量0.30 mL·min-1;进样量5μL。梯度洗脱程序:0~1.0 min时,A为70%;1.0~11.0 min时,A由70%降至6%;11.0~11.1 min时,A由6%跳转至0,保持2.9 min;14.0~14.1 min时,A由0跳转至70%,保 持2.9 min。
2)质谱条件 电喷雾离子源,正离子模式;离子传输管温度350℃,雾化器温度320℃;多反应监测模式扫描。其他质谱参数见表1和表2,其中,“*”为定量离子对。
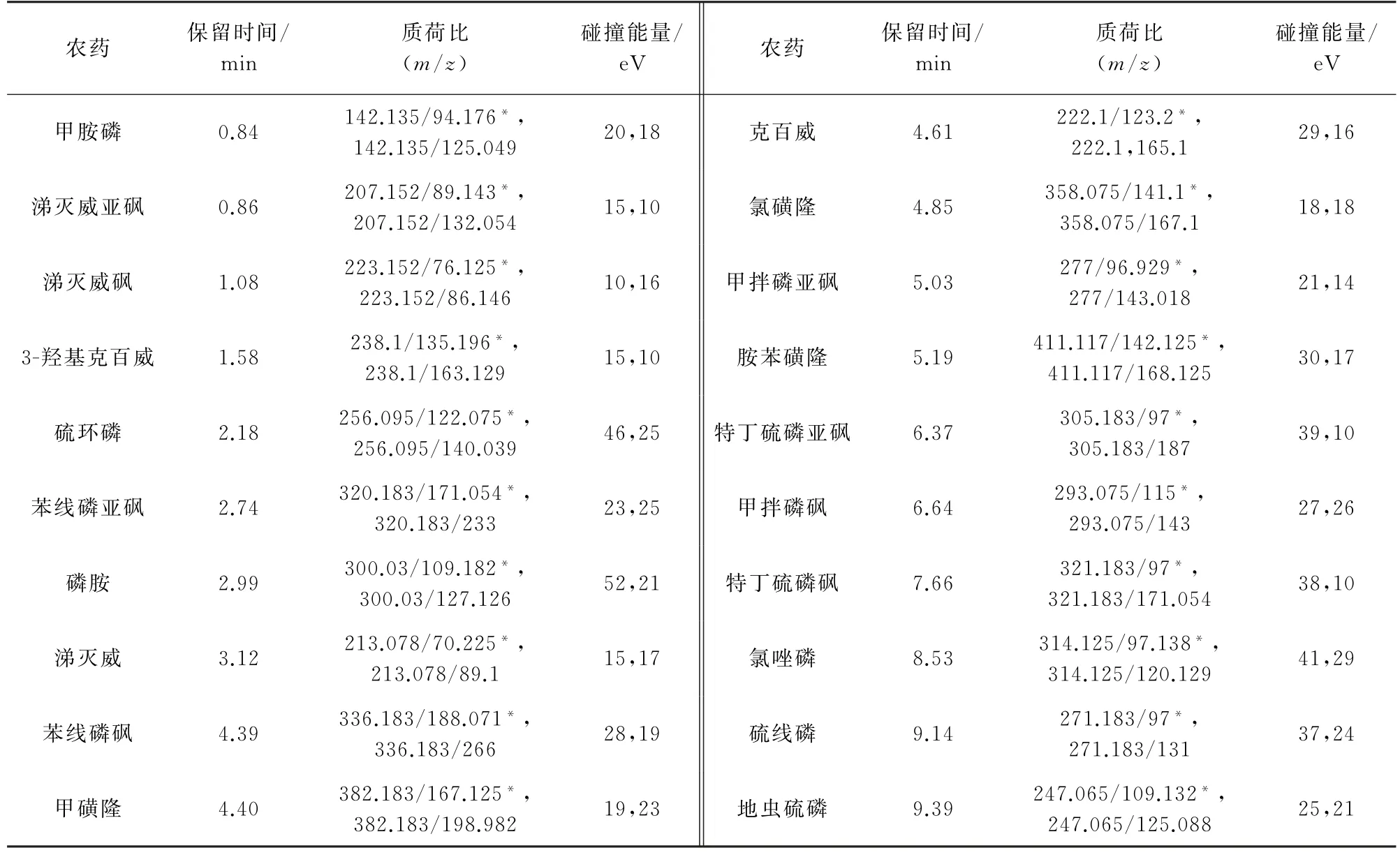
表1 部分农药的质谱参数(HPLC-MS/MS)Tab.1 MS parameters of some pesticides(HPLC-MS/MS)
1.2.2 GC-MS/MS
1)色谱条件 Agilent Technologies HP-5MS毛细管色谱柱(30 m×0.25 mm,0.25μm),载气为氦气(纯度不小于99.999%);流量1.2 mL·min-1;进样口温度250℃;进样量1μL,不分流进样。柱升温程序:初始温度为50℃,保持2 min;以50℃·min-1速率升温至150℃,再以5℃·min-1速率升温至200℃,最后以16℃·min-1速率升温至280℃,保持8 min。
2)质谱条件 电子轰击离子源,正离子模式;离子源温度300℃,接口温度280℃;多反应监测模式扫描。其他质谱参数见表2和表3,其中,“*”为定量离子对。
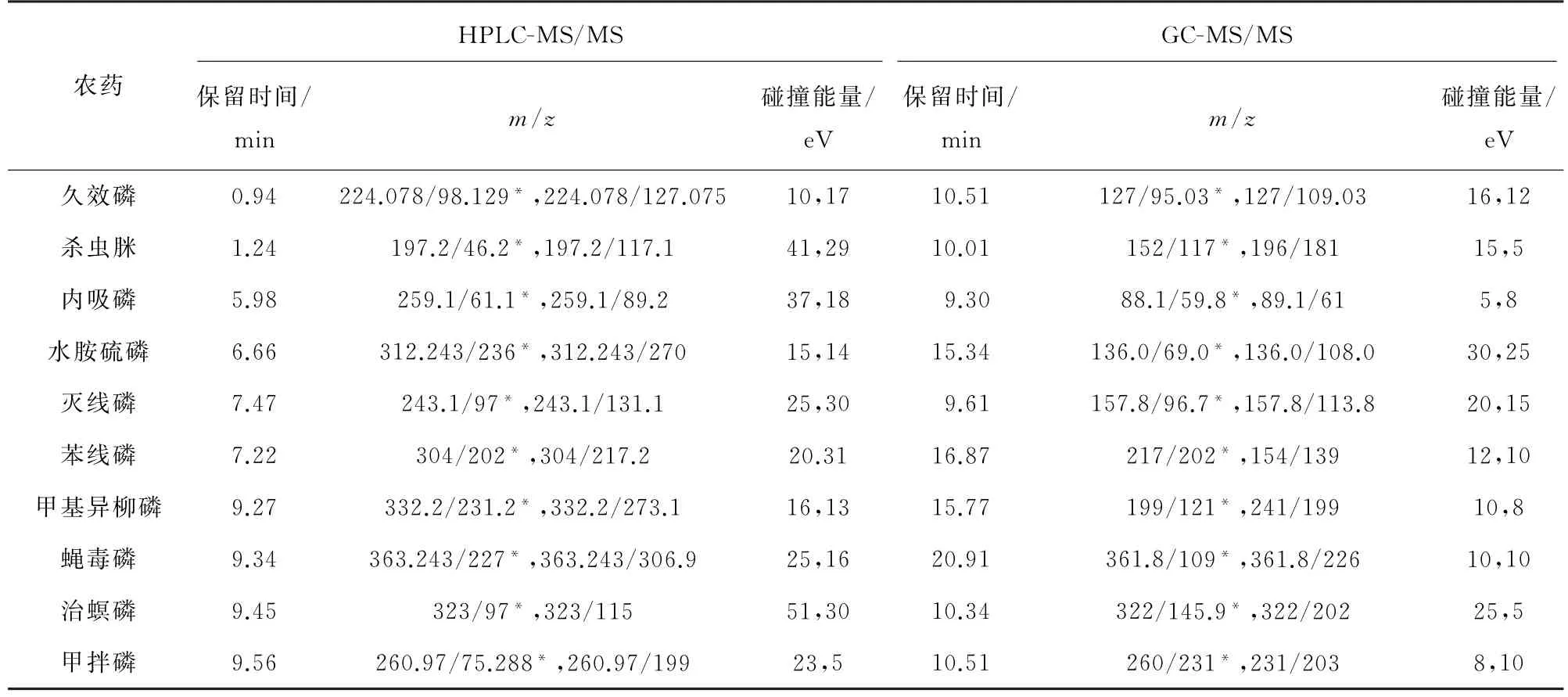
表2 部分农药的质谱参数(HPLC-MS/MS和GC-MS/MS)Tab.2 MS parameters of some pesticides(HPLC-MS/MS and GC-MS/MS)
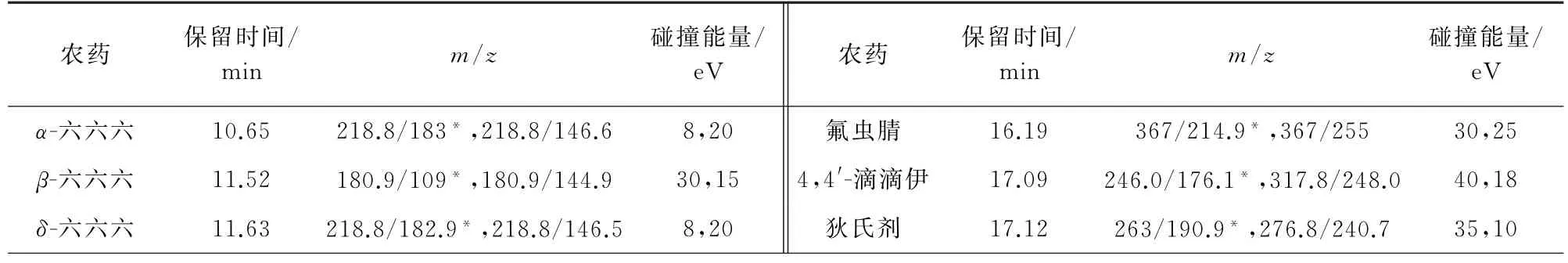
表3 部分农药的质谱参数(GC-MS/MS)Tab.3 MS parameters of some pesticides(GC-MS/MS)
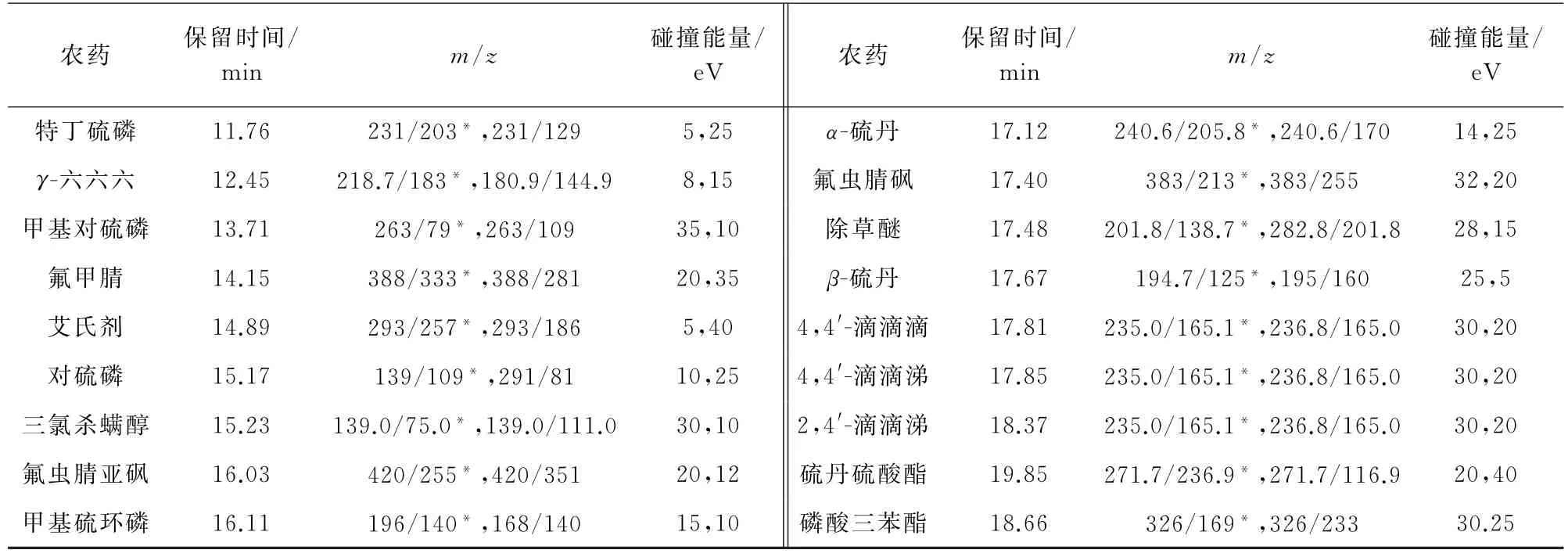
表3(续)
1.3 试验方法
1.3.1 样品前处理
1)固相萃取法
称取样品粉末5 g(精确至0.01 g)和氯化钠1 g,混匀,加入50 mL乙腈,以转速12 000 r·min-1均质2 min,以转速4 200 r·min-1离心5 min,取上清液于200 mL鸡心瓶中,沉淀中再加入乙腈50 mL,以转速12 000 r·min-1均质1 min,以转速4 200 r·min-1离心5 min,合并两次上清液,减压浓缩至1~3 mL,再用乙腈定容至10 mL,得到提取液。
在装有石墨化碳黑(GCB)的氨基复合固相萃取小柱(500 mg/6 mL)中加入约2 g的无水硫酸钠,用10 mL体积比为3∶1的乙腈-甲苯混合液淋洗固相萃取小柱,弃去流出液;下接25 mL离心管,取上述提取液4 mL于固相萃取小柱中,用25 mL体积比为3∶1的乙腈-甲苯混合液洗脱,将洗脱液置氮吹仪上,于40℃水浴浓缩至近干,加入0.02 mL内标溶液和2 mL乙腈溶解后,再用0.22μm有机滤膜过滤,收集滤液。
2)Qu ECh ERS法
称取样品粉末3 g(精确至0.01 g),置于50 mL聚苯乙烯具塞离心管中,加入1%(体积分数)冰乙酸溶液15 mL,涡旋使样品充分浸润,静置30 min,加入乙腈15 mL,涡旋混匀,剧烈振荡1 min,加入7.5 g质量比为4∶1的无水硫酸镁-无水乙酸钠混合粉末,混匀,再剧烈振荡1 min,于冰浴中冷却10 min,以转速4 200 r·min-1离心5 min,取上清液置于净化管[400 mgN-丙基乙二胺(PSA)/400 mg C18吸附剂/200 mg GCB/1 200 mg硫酸镁]中,涡旋混匀,剧烈振荡1 min,以转速4 200 r·min-1离心5 min,吸取上清液5 mL,置氮吹仪上于40℃水浴浓缩至近干,加入0.01 mL内标溶液,再用乙腈稀释至1 mL,涡旋混匀,得到待测样品溶液。
1.3.2 样品测定
按照仪器工作条件进行测定,HPLC-MS/MS采用外标法定量,GC-MS/MS采用内标法定量。六六六的含量以α-666、β-666、δ-666、γ-666的含量之和表示;滴滴涕的含量以4,4′-滴滴伊、4,4′-滴滴滴、4,4′-滴滴涕、2,4′-滴滴涕的含量之和表示;苯线磷的含量以苯线磷、苯线磷砜、苯线磷亚砜的含量之和表示;特丁硫磷的含量以特丁硫磷、特丁硫磷砜、特丁硫磷亚砜的含量之和表示;甲拌磷的含量以甲拌磷、甲拌磷砜、甲拌磷亚砜的含量之和表示;克百威的含量以克百威、3-羟基克百威的含量之和表示;涕灭威的含量以涕灭威、涕灭威砜、涕灭威亚砜的含量之和表示;硫丹的含量以α-硫丹、β-硫丹、硫丹硫酸酯的含量之和表示;氟虫腈的含量以氟虫腈、氟甲腈、氟虫腈砜、氟虫腈亚砜的含量之和表示。
2 结果与讨论
2.1 净化管的选择
Qu ECh ERS法中GCB可去除色素等非极性干扰物;PSA可吸附糖类、有机酸等极性干扰物;C18吸附剂可去除脂类、固醇等非极性杂质;硅胶则可除掉糖类等极性较大的杂质;硫酸镁可去除水分。填料越多,去除杂质的效果也越好,但是对目标物的吸附也越多。试验考察了3种净化管(250 mg PSA/35 mg GCB/750 mg硫酸镁/250 mg C18吸附剂;300 mg PSA/300 mg C18吸附剂/90 mg GCB/900 mg硫酸镁/300 mg硅胶;400 mg PSA/400 mg C18吸附剂/200 mg GCB/1 200 mg硫酸镁)对空白加标样品(加标量0.02 mg·kg-1)净化效果的影响。结果表明:样品经前两种净化管净化后,待测样品溶液颜色较深,检测时对仪器的伤害较大;填料为400 mg PSA/400 mg C18吸附剂/200 mg GCB/1 200 mg硫酸镁的净化管对大多数目标物的净化效果较好,各目标物的回收率均在55.0%~110%内。因此,QuECh ERS法中选用填料为400 mg PSA/400 mg C18吸附剂/200 mg GCB/1 200 mg硫酸镁的净化管来对样品进行前处理。
2.2 标准曲线、检出限和测定下限
移取适量的混合标准储备溶液和内标溶液,分别用甲醇、乙腈或丙酮逐级稀释,配制成各目标物质量浓度为5,10,20,50,100,200μg·L-1,内标质量浓度为1 mg·L-1的混合标准溶液系列,按照仪器工作条件对上述混合标准溶液系列进行测定。以各目标物的质量浓度为横坐标,对应的峰面积(HPLC-MS/MS)或目标物峰面积与内标峰面积的比值为纵坐标(GC-MS/MS)绘制标准曲线。结果显示,33种农药及其异构体或氧类似物标准曲线的线性范围为5~200μg·L-1(HPLC-MS/MS)或10~200μg·L-1(GC-MS/MS),线性参数见表4~表6。
对0.02 mg·kg-1空白加标样品进行测定,以信噪比为3和10时对应的质量分数为检出限和测定下限,结果见表4~表6。
表4 部分农药的线性参数、检出限和测定下限(HPLC-MS/MS)Tab.4 Linearity parameters,detection limits and lower limits of determination of some pesticides(HPLC-MS/MS)
表5 部分农药的线性参数、检出限和测定下限(GC-MS/MS)Tab.5 Linearity parameters,detection limits and lower limits of determination of some pesticides(GC-MS/MS)
表5(续)
表6 部分农药的线性参数、检出限和测定下限(HPLC-MS/MS和GC-MS/MS)Tab.6 Linearity parameters,detection limits and lower limits of determination of some pesticides(HPLC-MS/MS and GC-MS/MS)
2.3 精密度和回收试验
按照试验方法对阴性黄芩样品进行3个浓度水平的加标回收试验,每个浓度水平平行测定6次,计算回收率和测定值的RSD,结果见表7~表9。
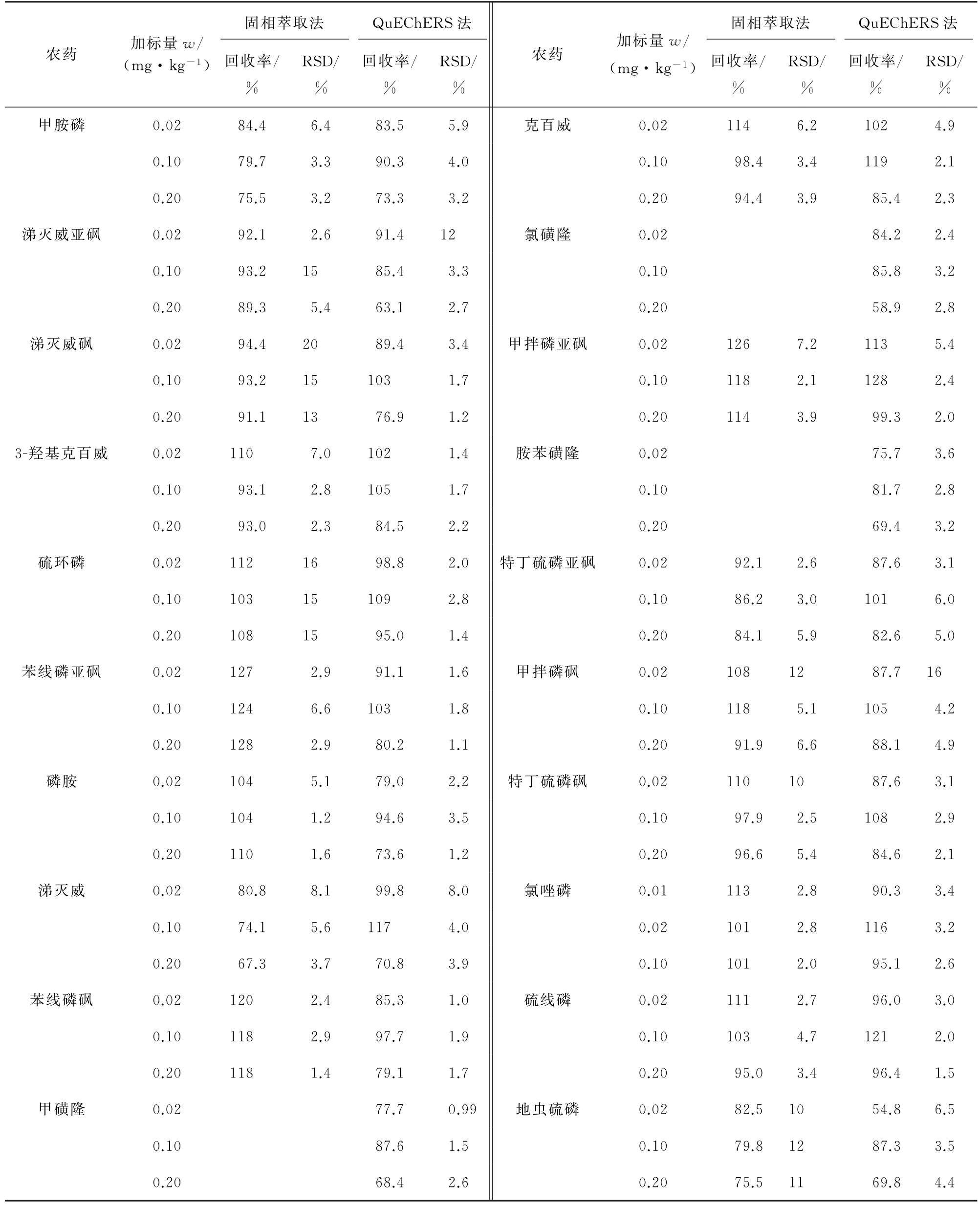
表7 两种前处理方式下部分农药的精密度和回收试验结果(HPLC-MS/MS,n=6)Tab.7 Results of tests for precision and recovery of some pesticides with two pretreatment methods(HPLC-MS/MS,n=6)
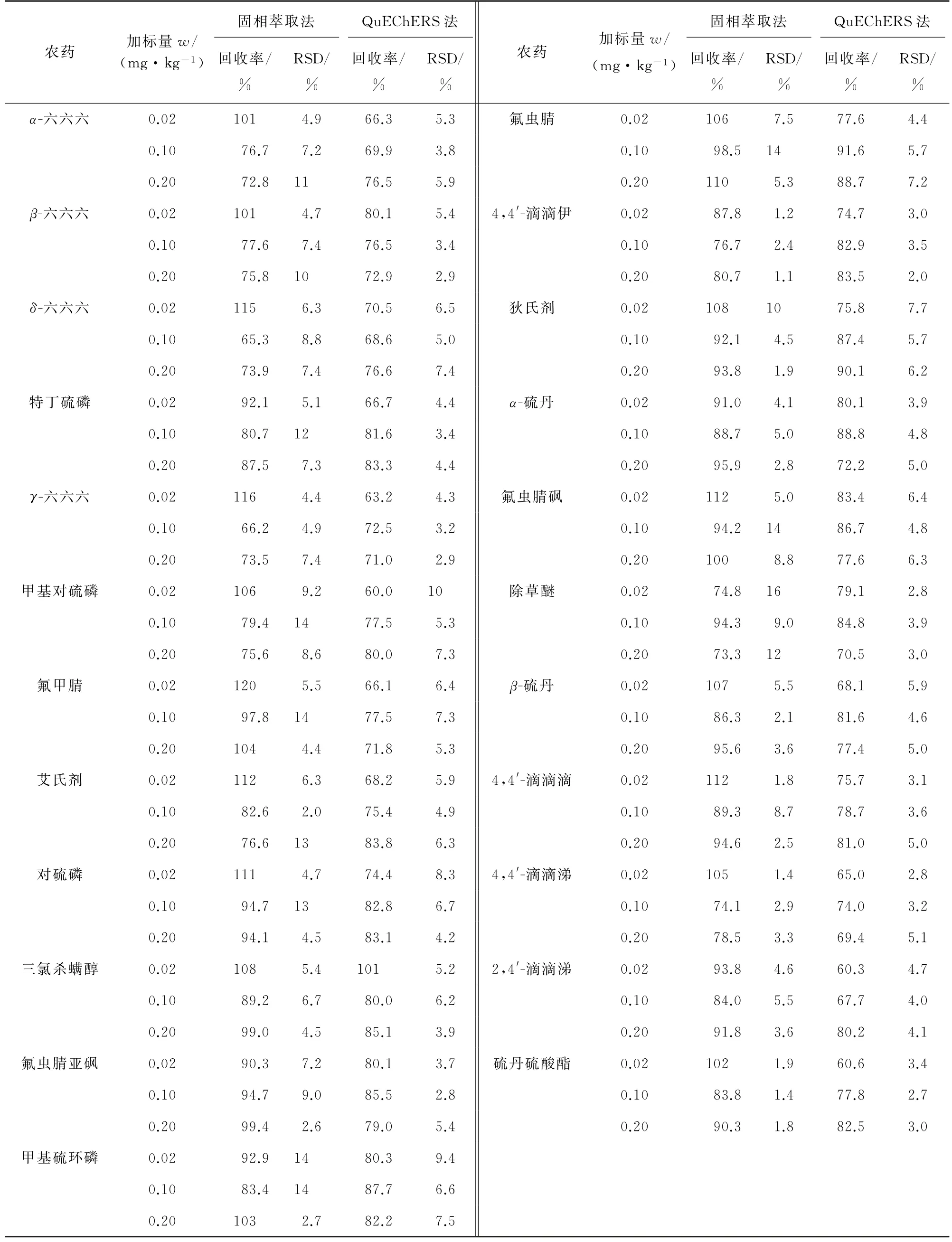
表8 两种前处理方式下部分农药的精密度和回收试验结果(GC-MS/MS,n=6)Tab.8 Results of tests for precision and recovery of some pesticides with two pretreatment methods(GC-MS/MS,n=6)
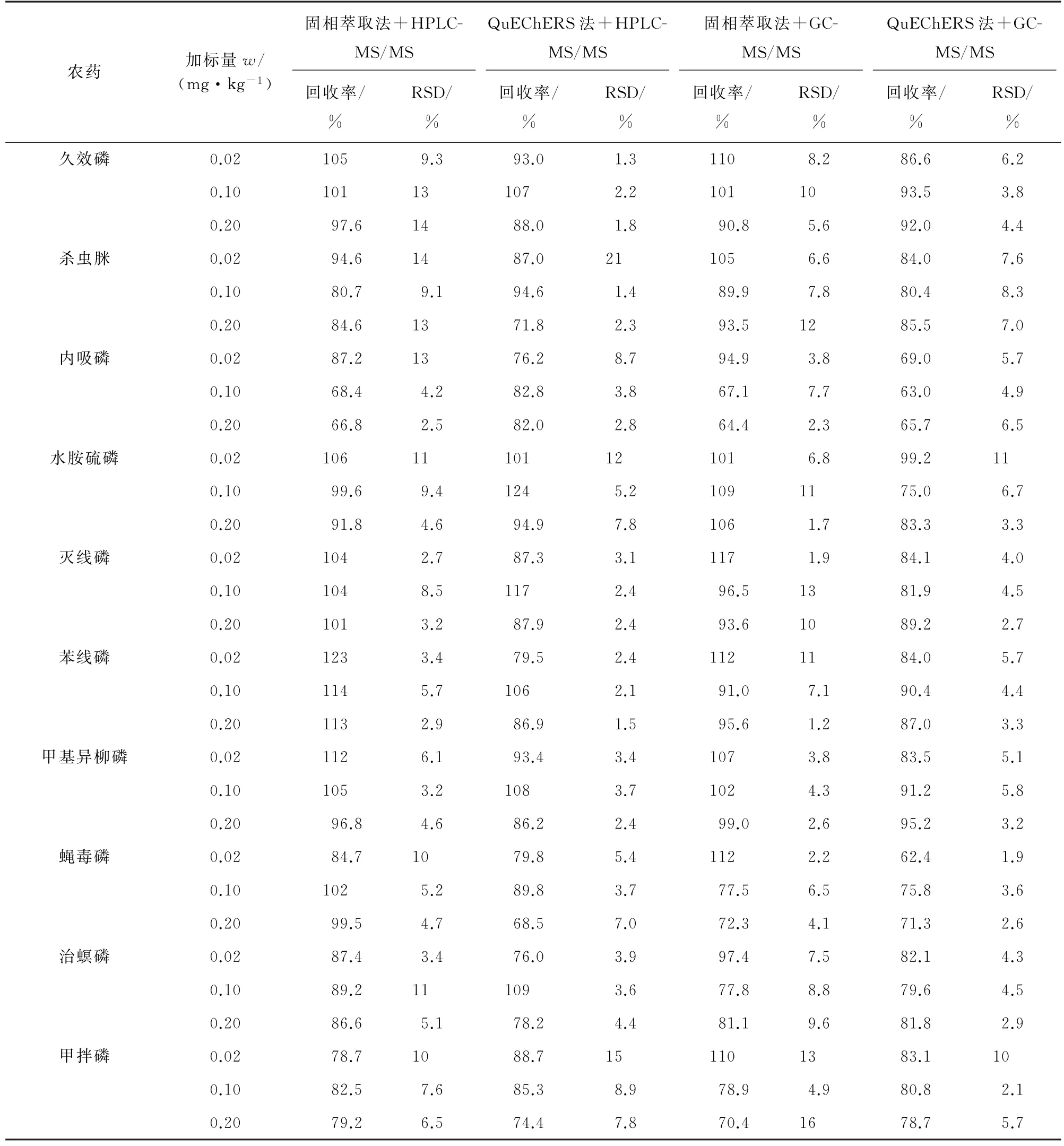
表9 两种前处理方式下部分农药的精密度和回收试验结果(HPLC-MS/MS和GC-MS/MS,n=6)Tab.9 Results of tests for precision and recovery of some pesticides with two pretreatment methods(HPLC-MS/MS and GC-MS/MS,n=6)
结果表明:两种前处理方式、两种检测方法下各目标物的回收率为54.8%~128%,测定值的RSD为0.99%~21%,表明方法准确可靠,可用于实际样品的分析检测。
2.4 样品分析
按照试验方法,对黄芩片、紫苏叶、黄苓、黄柏、小茴香、当归、半夏、山麦冬等中药材样品进行测定,结果见表10,“#”表示测定结果超出Ch P2020限值。除表10检出的物质外,其余农药均未检出。Ch P2020中涕灭威的限值为100μg·kg-1,甲胺磷、磷胺、克百威、水胺硫磷的限值为50μg·kg-1,硫环磷的限值为30μg·kg-1,灭线磷、硫线磷、甲基异柳磷、苯线磷、特丁硫磷、甲拌磷的限值为20μg·kg-1。
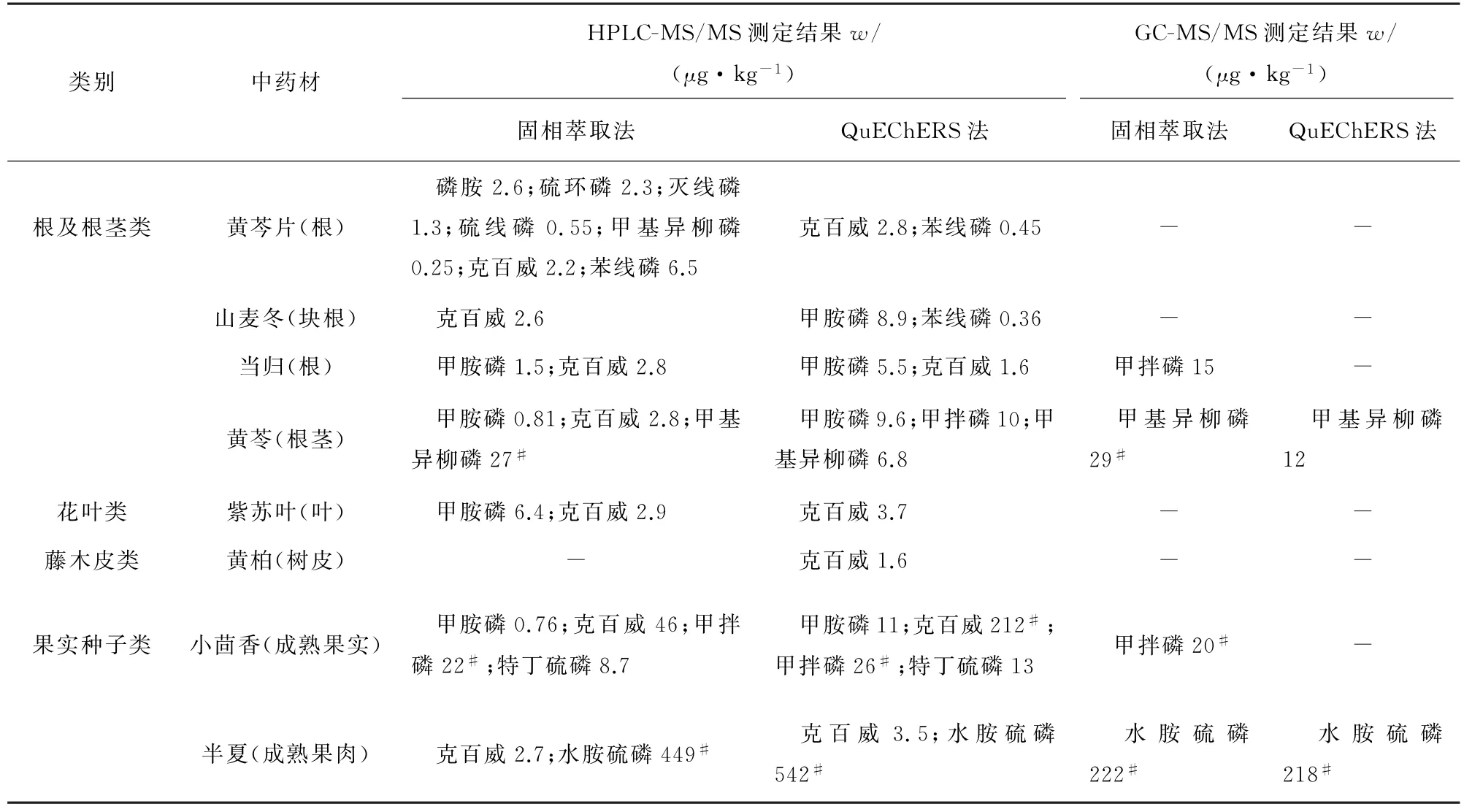
表10 样品分析结果Tab.10 Analytical results of the samples
由表10可以看出,目前所用到的农药主要为有机磷和氨基甲酸酯两类农药,且果实种子类和根及根茎类中药材中的部分农药含量超出Ch P2020限值。在检出的农药中,苯线磷及其两种代谢物苯线磷砜和苯线磷亚砜均有检出,甲拌磷和特丁硫磷只检出亚砜代谢物甲拌磷亚砜和特丁硫磷亚砜,氨基甲酸酯类农药中主要检出克百威。农业部公告199号[13]中已全面禁止甲胺磷、磷胺等19种高毒性农药在蔬菜、果树、茶叶、中草药材中的使用;农农发[2010]2号[14]中禁止克百威、甲基异柳磷用于蔬菜、果树、茶叶、中草药材,但仍有少量的甲胺磷及超出Ch P2020限值的克百威和甲基异柳磷被检出。一方面可能是土壤中含有该类农药残留,其在中药材的生长中发生富集导致;另一方面和一些农药母体的使用有关,如乙酰甲胺磷代谢产生甲胺磷,丁硫克百威代谢产生克百威,这两种药物于2017年农业部公告2552号[15]中被禁止使用。
2.4.1 固相萃取法和QuECh ERS法净化效果的比较
试验发现:采用氨基复合固相萃取小柱净化后,黄柏、小茴香和当归的提取液均呈现不同程度的黄色;而采用QuECh ERS法处理后,小茴香和当归样品溶液没有颜色,黄柏仍有黄色;紫苏叶经氨基复合固相萃取小柱净化后没有颜色,经Qu ECh ERS法净化后颜色较深,氨基复合固相萃取小柱对其余7种样品的净化效果优于Qu ECh ERS法的,但前者的处理时间较长,且试验中用到较多对环境极不友好的甲苯和乙腈。因此,紫苏叶在批量检测时建议采用氨基固相萃取小柱结合QuEChERS法处理,其余7种中药材样品建议直接采用Qu ECh ERS法处理。
2.4.2 同一物质测定结果的比较
超出Ch P2020限值的农药中,HPLC-MS/MS和GC-MS/MS均检出的物质有甲拌磷、水胺硫磷和甲基异柳磷。从检测方法来看,小茴香经固相萃取法和QuEChERS法处理后,甲拌磷在HPLCMS/MS上的检测结果无明显差异,而在GC-MS/MS上的检测结果差异较大,建议分析甲拌磷时以灵敏度更高的HPLC-MS/MS分析结果为准;采用HPLC-MS/MS和GC-MS/MS分析甲基异柳磷所得结果无明显差异,可根据实际情况选择仪器分析;水胺硫磷在HPLC-MS/MS上的检测结果约为GCMS/MS检测结果的2倍,并且试验发现水胺硫磷在HPLC-MS/MS中有较强的基质效应,因此实际分析中建议选用GC-MS/MS分析。
从前处理方法来看,固相萃取法的检出率相比Qu ECh ERS法的要高,样品经QuECh ERS法处理后存在误判的情况。如黄苓经两种方法处理后,甲基异柳磷的测定结果存在合格与不合格的差异,建议样品在QuEChERS法处理后,若甲基异柳磷检出结果大于0.01μg·kg-1但低于Ch P2020限值或者高于Ch P2020限值时,应采用固相萃取法处理复检;小茴香经固相萃取法、Qu ECh ERS法处理后,克百威的测定值差异较大,可能是QuEChERS法净化效果不佳导致目标物出现假阳性的结果。
本工作通过优化QuEChERS法中净化管的填料,分别采用固相萃取法和QuECh ERS法对8种中药材进行前处理,用HPLC-MS/MS和GC-MS/MS测定样品中33种禁用农药的含量,比较了不同前处理方法的净化效果,分析了不同前处理方法对同一农药检测结果的差异性、不同类别中药材中农药残留的检出情况以及不同检测方法对同一农药检测结果的影响,得出不同用药部位中药材宜采用的前处理方法和检测手段,可为中药材的农残分析检测提供参考。
