脾脏首发ALK阳性大B细胞淋巴瘤1例
2022-09-28李子瑶刘卫平
李子瑶,刘卫平
患者男性,51岁。体检发现脾脏单发占位,无症状。查体:脾脏大,余无明显异常。腹部增强CT示脾脏体积增大,实质强化欠均匀,内见一大小7.7 cm×6.2 cm稍低密度肿块影,边界较清,欠均匀轻度强化(图1)。PET-CT示脾脏体积增大,其内稍低密度肿块伴FDG摄取异常增高,提示恶性肿瘤可能性大,身体其他部位未见确切恶性病变征象。实验室检查仅发现乳酸脱氢酶(LDH)水平升高。HIV初筛阴性。

图1 CT示脾脏内见稍低密度肿块影,边界较清
病理检查眼观:脾脏大小14.5 cm×10.5 cm×6 cm,重470 g。被膜下见一灰白色肿物,肿物大小7 cm×6.5 cm×5 cm(图2),切面灰白、灰黄色,鱼肉样,质软。镜检:病变区内未见正常脾组织结构,为成片、灶性分布或窦性浸润的肿瘤组织所取代,伴多灶坏死或退行性改变,见凋亡小体。瘤细胞形态较一致,呈浆母细胞样,易见核分裂,可见异型多核瘤巨细胞(图3A)。免疫表型:瘤细胞CD45、MUM1、CD138(图3B)、CD30、CD45、EMA和ALK(图3C)均阳性,B细胞抗原(CD20、CD79a、PAX-5)、T细胞抗原(CD2、CD3、CD4、CD5、CD7、Granzyme B、TIA1)和组织细胞抗原(CD68、CD163)阴性,HHV8、CD38、Cyclin D1、BCL-2、MYC、S-100、HMB-45、desmin、SMA、Kappa和Lambda等均阴性。EBER1/2原位杂交检测为阴性。Ki-67增殖指数为80%。FISH检测ALK基因易位(图3D)。肿瘤组织二代测序检出CLTC-ALK基因融合。未见IgH、Igκ、TCRγ基因克隆性重排。
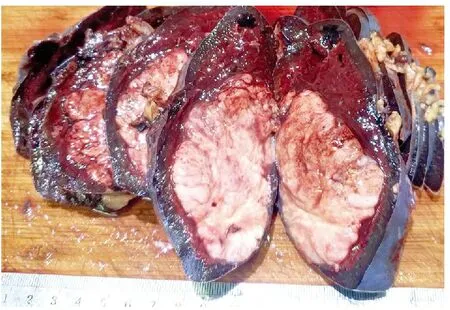
图2 脾脏被膜下可见一灰白色肿物,呈鱼肉样
病理诊断:非霍奇金淋巴瘤,ALK+大B细胞淋巴瘤(large B-cell lymphoma, LBCL),临床ⅣA期。患者接受5周期克唑替尼+CHOP化疗后,临床及影像学评估为完全缓解。随访17个月,现病情稳定。
讨论ALK+LBCL是一种罕见的高度侵袭性成熟B细胞淋巴瘤,该肿瘤于1997年由Delsol等[1]首次报道,在WHO(2008)造血和淋巴组织肿瘤分类中被列为一种独立疾病[2]。ALK+LBCL占LBCL比例<1%,目前国内外文献报道的ALK+LBCL有200余例[3-4]。
ALK+LBCL多发生于免疫功能正常人群,男女比为4 ∶1,中位发病年龄38岁(年龄范围9~90岁)。好发于淋巴结,病变也可累及结外器官或组织,如鼻咽、胃肠道、皮肤和软组织等[5-6],其中文献报道的脾脏受累者合计7例,未见脾脏首发病例。多数患者呈侵袭性临床过程:患者发病时已为晚期(Ⅲ/Ⅳ期),伴B症状(发热、盗汗、体重减轻)、骨髓浸润、LDH升高。本例为免疫功能正常的中年男性患者,体检发现脾脏单发占位,实验室检查仅LDH水平升高,身体其他部位均未见累及。
组织病理学和分子遗传学:受累组织结构部分或完全破坏。肿瘤细胞呈弥漫或窦性浸润模式,也可呈巢或片状生长。瘤细胞形态单一,多为免疫母细胞样细胞或浆母细胞样细胞。部分病例可见多核瘤巨细胞。肿瘤细胞表达浆细胞相关抗原,如CD38、CD138、MUM1和VS38c,不表达B或T细胞相关抗原,如CD20、CD79a、CD3和CD5;部分表达EMA和CD45,常表达IgA。HHV8为阴性。EBER1/2原位杂交检测为阴性。瘤细胞强表达ALK蛋白,最常见的表达模式是胞质内颗粒状阳性。肿瘤有特征性ALK基因重排,最常见的遗传学改变是t(2; 17)(p23; q23)产生CLTC-ALK融合。ALK+LBCL也存在其他遗传性改变,如:NPM-ALK、SQSTM1-ALK、SEC31A-ALK、RANBP2-ALK和IGL-ALK等。本例形态学、免疫表型和分子遗传学改变均支持为ALK+LBCL。
鉴别诊断:(1)ALK+LBCL首先需与具有浆母细胞形态或分化特征的一组淋巴造血组织肿瘤相鉴别[7],包括:浆母细胞性淋巴瘤(plasmablastic lymphoma, PBL)、原发渗出性淋巴瘤(primary effusion lymphoma, PEL)、浆母细胞性浆细胞瘤/浆细胞骨髓瘤(plasmablastic plasma cell myeloma/plasmacytoma)、HHV8+弥漫大B细胞淋巴瘤,非特指(human herpesvirus 8-positive diffuse large B-cell lymphoma, not otherwise specified, HHV8+DLBCL, NOS)和HHV8+多中心Castleman病。PBL、PEL及HHV8+DLBCL, NOS常见于HIV感染或其他免疫功能缺陷的患者。需注意患者年龄和机体免疫功能状态、病变部位,是否表达ALK蛋白或存在ALK重排。(2)ALK+LBCL还需与其他表达ALK蛋白的肿瘤相鉴别,包括:ALK阳性间变性大细胞淋巴瘤(anaplastic large cell lymphoma, ALK-positive, ALK+ALCL)、ALK阳性非小细胞肺癌(non-small cell lung cancer, ALK-positive, ALK+NSCLC)、炎性肌纤维母细胞肿瘤(inflammatory myofibroblastic tumor, IMT)和ALK阳性组织细胞增生性疾病/肿瘤。可结合病变部位、病理形态、免疫表型、ALK伙伴基因,以及临床及影像学检查进行综合分析。如在ALK+ALCL中最常见的是NPM-ALK融合;在ALK阳性肺腺癌中最常见的是EML4-ALK融合;在IMT中最常见的是TPM3/4-ALK融合;在ALK阳性组织细胞增生症中最常见的是KIF5B-ALK融合。(3)ALK+LBCL还要与其他非淋巴造血组织肿瘤鉴别,如黑色素瘤、低分化癌及小圆细胞恶性肿瘤等。结合临床表现、组织病理学、免疫表型及分子遗传学有助于疾病的诊断。
ALK+LBCL呈高度侵袭性临床过程,大多数患者对CHOP标准治疗或CHOP衍生治疗方案效果不佳,复发率高,进展迅速,预后差。多数患者在确诊2年内死亡,中位生存期为1.83年。在Ⅲ/Ⅳ期患者中,5年总生存期仅为8%。克唑替尼是一种ALK激酶抑制剂,对复发难治ALK+LBCL有一定的疗效[8-9]。本例患者发病时即为ⅣA期,但临床过程呈惰性,予5周期克唑替尼+CHOP方案治疗,治疗反应佳,目前为无瘤生存,继续随访中。
本例为脾脏首发且身体其他部位均未见肿瘤累及的ALK+LBCL,国内外文献尚未见报道。ALK+LBCL是一种罕见的侵袭性淋巴瘤。多见于免疫功能正常中年人。常好发于淋巴结。瘤细胞常呈免疫母细胞样或浆母细胞样,呈弥漫性或窦性浸润模式。肿瘤细胞不表达B或T细胞相关抗原,表达CD38、CD138、MUM1、BOB1及OCT2等。诊断时应注意与其他疾病相鉴别。特征性ALK蛋白表达及CLTC-ALK融合是其重要的诊断依据。患者常采用CHOP+ALK激酶抑制剂进行治疗,多数患者预后不佳。
