高效液相色谱法同时测定氢达乳膏中三组分含量
2022-09-02丁志军李小兰付志媛
丁志军,李小兰,付志媛
1.江西省皮肤病专科医院,江西 南昌 330001;2.江西省皮肤病临床医学研究中心,江西 南昌 330001;3.国家皮肤与免疫疾病临床医学研究中心分中心建设单位,江西 南昌 330001
氢达乳膏是由盐酸达克罗宁、羟苯乙酯、氢化可的松、白凡士林、平平加A-20、枸橼酸、甘油、水等组成的水包油型乳膏剂,收录于2020 年版《江西省医疗机构制剂规程》,具有止痛、止痒作用,主要用于皮炎、带状疱疹后遗神经痛等,疗效确定[1]。原标准中制剂含量测定项采用氯化三苯四氮唑比色法[2]控制氢化可的松用量,不仅方法专属性差,而且样品处理步骤复杂繁琐、操作过程十分耗时,导致试验结果不理想,准确性不好、重现性差。对抑菌剂羟苯乙酯未建立质量控制方法。文献报道[3-4],羟苯乙酯用量应达到抑菌有效量;同时其有效浓度应不超过对人体的有害浓度。为此,本文对氢达乳膏中盐酸达克罗宁、羟苯乙酯、氢化可的松3 种主要成分的含量测定方法进行了研究。
1 材料
1.1 仪器
Agilent 1260 Infinity Ⅱ高效液相色谱仪(美国Agilent 公司,DAD 检测器);BT-125D Sartorius 电子天平(德国);UV-1800 紫外分光光度计(日本岛津公司)。
1.2 试药
盐酸达克罗宁对照品(中国食品药品检定研究院,批号100423-201102,含量99.8%);羟苯乙酯对照品(中国食品药品检定研究院,批号100847-201604,含量99.9%);氢化可的松对照品(中国食品药品检定研究院,批号100152-201707,含量99.2%);甲醇(色谱纯,Merck 公司);水(重蒸馏水,江西省皮肤病专科医院制剂室);其余试剂均为分析纯;氢达乳膏(江西省皮肤病专科医院制剂室,批号210201、210301、210401)。
2 方法与结果
2.1 色谱条件
采用Agilent C18 柱(250 mm×4.6 mm,5 µm)色谱柱;以枸橼酸-磷酸氢二钠缓冲液(含0.2%枸橼酸和0.2%磷酸氢二钠)-甲醇(34∶66)为流动相;流速为0.6 mL/min;柱温为25 ℃;检定波长:256 nm;进样体积为5 µL。理论塔板数按盐酸达克罗宁峰计应不低于3 000。
2.2 溶液的制备
2.2.1 对照品溶液的制备分别取盐酸达克罗宁对照品、羟苯乙酯对照品、氢化可的松对照品适量,精密称定,加枸橼酸乙醇液(含0.12‰枸橼酸)溶解并稀释制成每1 mL 分别含61.920、6.044、60.288 µg的对照品溶液,即得。
2.2.2 供试品溶液的制备取样品适量(约相当于盐酸达克罗宁3 mg、羟苯乙酯0.3 mg、氢化可的松3 mg),精密称定,放置于50 mL 容量瓶中,加入一定量95%乙醇,放置于70 ℃水浴中加热大约5 min,振动摇晃量瓶使乳膏中盐酸达克罗宁、羟苯乙酯及氢化可的松溶解,放凉至室温后,用95%乙醇溶液稀释并定容至刻度,摇匀,放置于冰箱中冷藏2 h以上,取出,用滤纸快速滤过,收集续滤液放至室温,即得。
2.2.3 阴性对照溶液的制备取按氢达乳膏处方工艺方法制备的缺盐酸达克罗宁、羟苯乙酯、氢化可的松的阴性样品适量,按上述“2.2.2”项下方法要求同法制备阴性对照溶液,即得。
2.3 专属性试验
分别精密吸取按对照品溶液、供试品溶液和阴性对照溶液各5 µL 注入液相色谱仪,采用上述“2.1”项下色谱条件,记录色谱图。结果,供试品色谱图在与对照品色谱图相应的位置上有相同的盐酸达克罗宁峰、羟苯乙酯峰、氢化可的松峰,而阴性对照色谱图在相应位置上无色谱峰干扰。色谱图见图1。
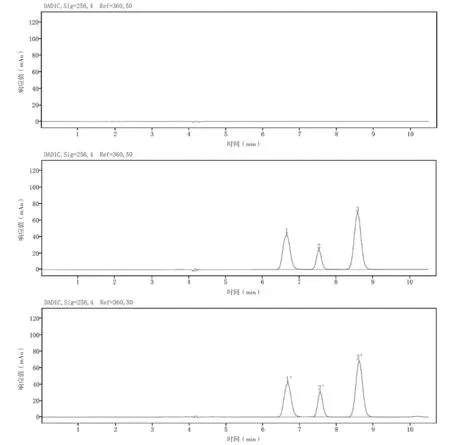
图1 高效液相色谱图:A.阴性对照溶液;B.对照品溶液;C.供试品溶液
2.4 线性关系考察
精密称取氢化可的松对照品18.84 mg、盐酸达克罗宁对照品19.35 mg,置25 mL 量瓶中,加含0.12‰枸橼酸乙醇溶液溶解并稀释至刻度,摇匀,得母液Ⅰ;取羟苯乙酯对照品15.11 mg,精密称定,置200 mL 量瓶中,加含0.12‰枸橼酸乙醇溶液溶解后,再用该乙醇溶液稀释至刻度,摇匀,得母液Ⅱ;精密吸取母液Ⅰ 1 mL、2 mL、3 mL、3 mL、5 mL,分别置25 mL、25 mL、25 mL、50 mL、50 mL 量瓶中,再精密吸取母液Ⅱ 1 mL、2 mL、3 mL、3 mL、5 mL,分别置上述量瓶中,加含0.12‰枸橼酸乙醇溶液稀释至刻度,摇匀,即得,采用上述“2.1”项下规定的色谱条件,分别精密吸取各溶液5 µL,注入高效液相色谱仪,记录色谱图,测定各峰峰面积,以各峰峰面积为纵坐标,各溶液进样量(µg)为横坐标,进行线性回归,盐酸达克罗宁的回归方程为:Y=1 869.896 6X+2.830 0,线性范围为0.155~0.464 µg(r=0.999 8);羟苯乙酯的回归方程为:Y=8 944.531 2X+2.124 8,线性范围为0.015~0.045 µg(r=0.999 8);氢化可的松的回归方程为:Y=2 989.251 6X+5.500 0,线性范围为0.151~0.452 µg(r=0.999 8)。
2.5 检出限
取盐酸达克罗宁对照品、羟苯乙酯、氢化可的松对照品适量,用枸橼酸乙醇液(含0.12‰枸橼酸)溶解并逐级稀释后测定,盐酸达克罗宁、羟苯乙酯、氢化可的松的检出限(3 S/N)分别为0.396 ng、0.058 ng、0.235 ng。
2.6 精密度试验
取对照品溶液,采用上述“2.1”项下规定的色谱条件,注入高效液相色谱仪,同法连续进样6 次,测得盐酸达克罗宁、羟苯乙酯、氢化可的松平均峰面积值,RSD 分别为0.96%、0.78%、1.01%,结果表明仪器精密度良好。
2.7 稳定性试验
精密吸取对照品溶液,分别在0、2、4、6、8 h采用上述“2.1”项下色谱条件进样,进样量为5 µL,测得盐酸达克罗宁、羟苯乙酯、氢化可的松平均峰面积值,RSD 分别为0.66%、0.87%和0.92%,结果表明盐酸达克罗宁、羟苯乙酯、氢化可的松在8 h 内稳定。
2.8 重复性试验
取样品(批号210201)6 份,采用万分之一分析天平分别精密称定重量,按上述“2.2.2”项下方法要求同法制备供试品溶液,采用上述“2.1”项下色谱条件进样,记录色谱图,测得各峰峰面积值,分别计算制剂氢达乳膏中盐酸达克罗宁、羟苯乙酯、氢化可的松含量,RSD 分别为0.92%、0.83%、0.58%,结果表明,此法重复性良好。
2.9 加样回收率试验
取已知含量的同一批样品(批号210201)6 份,精密称定,分别精密加入一定量的羟苯乙酯、氢化可的松、盐酸达克罗宁对照品,采用“2.2.2”项下方法制备供试品溶液,按上述“2.1”项下色谱条件进样测定,进样量为5 µL。结果见表1。
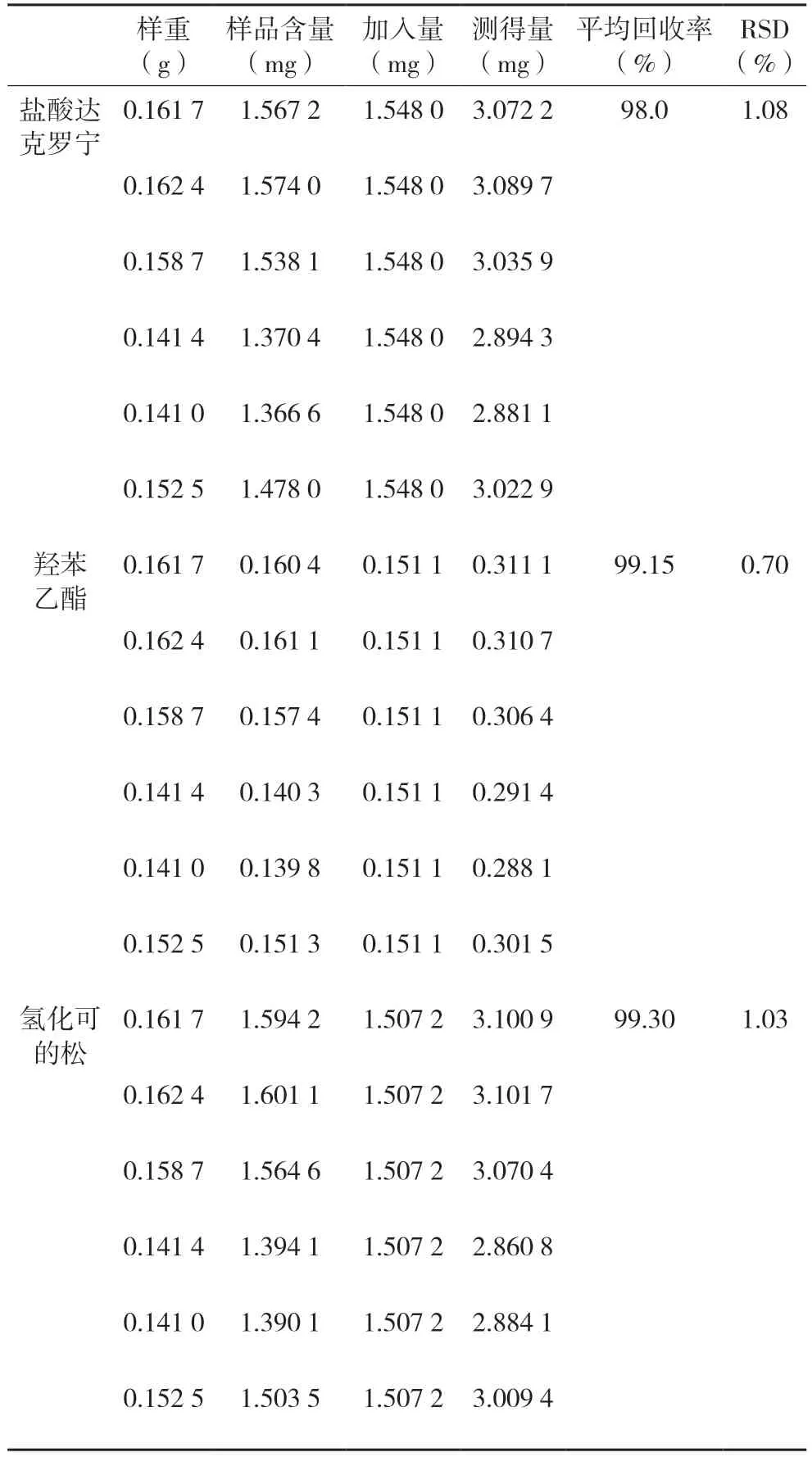
表1 加样回收率试验结果
2.10 样品含量测定
取样品3 批,按“2.2.2”项下方法制备供试品溶液,采用上述“2.1”项下色谱条件测定,按外标法计算氢达乳膏中盐酸达克罗宁、羟苯乙酯、氢化可的松标示量的百分含量,结果见表2。
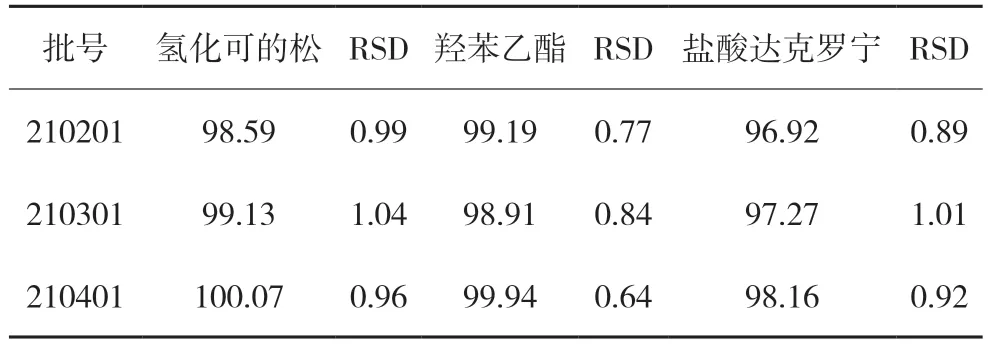
表2 样品含量测定结果(%)
3 讨论
3.1 流动相的选择
比较了含三乙胺的醋酸-醋酸铵缓冲液-甲醇[5]、枸橼酸-磷酸氢二钠缓冲液-甲醇[6]两种流动相体系,结果醋酸-醋酸铵缓冲液(含三乙胺)-甲醇作为流动相时,三乙胺的加入虽对盐酸达克罗宁峰拖尾现象有所改善,但同时液相色谱柱的柱效也下降明显。而以枸橼酸-磷酸氢二钠缓冲液(含0.2%枸橼酸和0.2%磷酸氢二钠)-甲醇(34∶66)为流动相时,盐酸达克罗宁、羟苯乙酯、氢化可的松色谱峰峰形对称(拖尾因子在0.90~1.10之间),分离度良好,柱效均较高。故流动相最终选择枸橼酸-磷酸氢二钠缓冲液-甲醇体系。
3.2 检测波长的选择
取盐酸达克罗宁、羟苯乙酯、氢化可的松混合对照品溶液5 μL,注入液相色谱仪(DAD 检测器),采集在190~400 nm 波长范围内光谱,从盐酸达克罗宁、羟苯乙酯和氢化可的松的最大吸收波长分别为282、256、244 nm,因处方中盐酸达克罗宁、羟苯乙酯和氢化可的松的含量比为10∶1∶10,为了兼顾三者的检测灵敏度,经试验摸索,最终选定测定波长为256 nm。
3.3 溶剂、提取温度及提取时间的考察
盐酸达克罗宁可溶于乙醇,略溶水中[7];羟苯乙酯易溶于甲醇、乙醇,几乎不溶于水中;氢化可的松略溶于乙醇,不溶于水中[8-9]。据此,在试验之初,我们以乙醇为溶剂制备对照品溶液及样品溶液,但实验过程中发现对照品溶液中盐酸达克罗宁含量下降较快,这与文献报道一致[10-11],因此,我们尝试在乙醇中加入酸以增加盐酸达克罗宁的稳定性,试验发现枸橼酸乙醇溶液(含0.12‰枸橼酸)能显著增加其稳定性,因此,选择枸橼酸乙醇液(含0.12‰枸橼酸)为溶剂制备对照品溶液。同时我们也对样品溶液制备时的提取温度、提取时间进行了单因素试验考察,发现水浴温度为70 ℃时,提取5 min 能将乳膏中盐酸达克罗宁、羟苯乙酯、氢化可的松提取完全。
