草酸铈和草酸铀的热分解机理及煅烧遗传性
2022-09-01吴纬洲李传博郑卫芳晏太红
吴纬洲,王 博,李传博,左 臣,郑卫芳,晏太红
中国原子能科学研究院 放射化学研究所,北京 102413
在核燃料后处理过程中,通常采用草酸沉淀法将溶液态硝酸钚转变为固态草酸钚沉淀,草酸钚经煅烧制备二氧化钚,用于MOX燃料制造[1]。制造MOX燃料对二氧化钚的产品粒径有严格的要求,粒径过大影响后续球磨工艺,过小则会产生可吸入颗粒[2]。此外,工业上对氧化物的球磨工艺并不能完全消除煅烧后氧化物的形貌特征[3],因此研究草酸盐热分解机制和煅烧前后颗粒形貌尺寸的变化关系从而有效控制形貌和粒径,对于钚尾端沉淀煅烧工艺改进具有重要意义。
镧、钇、钕、铈等稀土元素的草酸盐热分解机理一直是研究的重点[4-8],并能为锕系元素草酸盐研究提供参照,de Almeida等[9]就互补表征研究了钕、铈和钚(Ⅲ)草酸盐的热分解机理,发现草酸盐分解一般是脱结晶水形成不定形草酸盐后再进一步分解。Çlg等[10]发现不定形无水草酸铀的进一步热分解会先生成中间产物,氩气气氛的中间产物为UO1.5(CO3)0.5,而空气气氛的则还包括UO(CO3)。Vigier等[11]发现四价草酸钚在静态空气气氛下煅烧会还原生成混合草酸-碳酸钚(Ⅲ)中间物,而不是无结晶水的不定形草酸钚。
煅烧过程对最终氧化物粒径形貌等性质有较大的影响。柳召刚等[12]发现煅烧温度升温速率过快会导致氧化铈晶面破碎从而导致粒度分布变宽,高温则会导致颗粒松散密度升高。康秋珍等[13]的研究发现,分步灼烧和延长灼烧时间有利于获得大粒度氧化铈。张亚文等[14-16]发现短时间的灼烧能够有利于其颗粒分散,但长时间的灼烧下,颗粒会因为高温烧结形成团聚体,从而导致粒径的增大。Orr等[17]发现煅烧温度上升,二氧化钚产品的微晶尺度增加,但颗粒粒度的质量分布却对此并不敏感。
草酸盐煅烧后形貌基本得以保留,粒度则有一定程度的减小。马莹等[18]的氧化钇制备实验、陈志刚等[19]的球状、片状、棒状草酸铈煅烧实验、Duvieubourg-Garela等[20]的方形片状四价草酸铀煅烧实验、de Almeida等[9]的草酸钚煅烧实验均表明草酸盐前驱体形貌在煅烧后依然得以保留。马莹等[18]的氧化钇制备实验和Mulford等[3]的草酸钚粒度研究则表明煅烧后颗粒粒度有一定程度上的减小。
本工作选用铈作为钚的替代元素,一方面是由于镧系元素与锕系元素有类似的结构,经常作为锕系元素的非放研究替代物,另一方面是铈和钚的草酸盐在空气中热分解最终产物均是四价氧化物。此外,铀与钚同为锕系元素,其草酸盐化合价同为四价,但铀的价态相比于铈更加复杂,草酸铀在不同气氛下的煅烧热分解会得到不同价态的氧化物,因此对草酸铀热分解过程的研究需要在空气和氩气不同的气氛下进行。针对草酸盐的热分解机理和煅烧前后颗粒形貌尺寸遗传性问题,本工作拟采用同步热分析法(TG/DSC)开展草酸铈、草酸铀热分解反应的研究,以获取热分解过程的数据,并拟用X射线衍射仪(XRD)辅助确定中间产物,结合文献结果阐释热分解机理,并对静态空气和氩气气氛下草酸盐前驱体煅烧前后尺寸形貌进行表征,以对钚尾端沉淀煅烧工艺的改进提供参考依据。
1 实验部分
1.1 仪器和试剂
C-MAG HS 7磁力搅拌加热器,德国IKA艾卡集团;YZ1515x/BT600FC蠕动泵,保定创锐泵业有限公司;VOS-310C真空干燥箱,上海爱朗仪器有限公司;SHB-Ⅲ真空泵,郑州长城科工贸有限公司;TGA/DSC3+热重分析仪,瑞士梅特勒-托利多公司;Bettersize2600E激光粒度仪,丹东百特仪器有限公司;QICPIC激光粒度仪,德国新帕泰SYMPATEC公司;D8Advance X射线衍射仪,布鲁克科技有限公司;VEGA3LMU扫描电子显微镜,泰斯肯贸易有限公司;SX-G07103马弗炉、SK-G06183管式炉,天津市中环实验电炉有限公司。
硝酸铈六水合物,分析纯,上海麦克林生化科技有限公司;硝酸,分析纯,上海阿拉丁生化科技股份有限公司;二水合草酸、水合肼(体积分数85%),分析纯,国药集团化学试剂有限公司;硝酸铀(Ⅳ)储备溶液,实验室用硝酸铀酰还原制备,溶液含水合肼维持还原氛围,含硝酸维持酸度,保存于冰箱中。
1.2 草酸铈和草酸铀沉淀颗粒的制备
草酸沉淀法批式反应器示意图示于图1。如图1所示,两股反应料液分别是硝酸盐料液与草酸料液,其中硝酸铈料液中使用分析纯的Ce(NO3)3配制,硝酸铀料液使用实验室硝酸铀(Ⅳ)储备溶液配制,加入水合肼和硝酸,控制酸度和还原氛围,保证四价铀不被氧化。用磁力加热搅拌器设定温度和搅拌速率,通过将对应的两股反应料液在烧杯中进行搅拌混合获得沉淀,再进行抽滤。抽滤时用去离子水反复洗涤样品,之后用真空干燥箱在60 ℃下干燥1 d,得到不同粒度的草酸盐颗粒。

图1 草酸沉淀法实验装置简图Fig.1 Experimental equipment diagram of oxalic acid precipitation method
1.3 同步热分析实验
草酸铈和草酸铀的同步热分析实验在TGA/DSC3+热重分析仪上进行,设置气氛为氩气或流动空气(20 mL/min),根据样品设置不同的升温范围,升温速率设为10 ℃/min。
1.4 热分解实验
热分解实验根据气氛不同选用不同的加热设备,静态空气气氛选用马弗炉,氩气气氛选用管式炉。
热分解实验主要为后续的遗传性研究实验和XRD验证中间产物实验提供样品:配合遗传性研究时,需要将草酸盐前驱体样品编号后同时进行加热煅烧;配合热分析曲线验证中间产物时,加热煅烧单一样品即可。
1.5 XRD表征实验
XRD表征实验在X射线衍射仪上进行。
1.6 颗粒表征
实验中采用激光粒度仪表征颗粒粒度,两台激光粒度仪均为湿法测试仪器。使用时将样品分散到水中,采用机械搅拌和机器外超声3 min的方式进行分散和去除软团聚,保证样品测试的准确和重复。测试完成后能够得到粒径的分布图和数据,包括D10、D50、D90(D10、D50、D90为样品累计粒度分布数分别达到10%、50%、90%时所对应的粒径)等,采用多次测量结果的平均值方便后续进行分析。
实验中采用扫描电子显微镜表征其形貌,将样品黏着在导电胶上送入电子显微镜样品室,能够获得具有一定分辨率、保真度高、真实的三维效应的样品图。
2 结果和讨论
2.1 空气气氛下Ce2(C2O4)3·9H2O热分解机理
利用热重分析仪考察了Ce2(C2O4)3·9H2O样品在流动空气气氛(20 mL/min)中以10 ℃/min速率升温的热分解过程,热分析曲线示于图2。

图2 Ce2(C2O4)3·9H2O在流动空气(20 mL/min)气氛中以10 ℃/min速率升温的热分解过程Fig.2 Thermal decomposition of Ce2(C2O4)3·9H2O on heating at 10 ℃/min in streaming air(20 mL/min)
利用马弗炉获得静态空气气氛下Ce2(C2O4)3·9H2O在200、400 ℃下的退火反应产物,并将其与草酸铈前驱体一同进行XRD表征,并分别与Ce2(C2O4)3·9H2O的标准卡片PDF#20-0268、Ce2(C2O4)3的标准卡片PDF#40-0570、CeO2的标准卡片PDF#34-0394对比,结果示于图3。

(a)——Ce2(C2O4)3·9H2O,(b)——Ce2(C2O4)3,(c)——CeO2图3 静态空气气氛下草酸铈前驱体(a)与200 ℃(b)、400 ℃(c)下退火反应产物的XRD图Fig.3 XRD spectrum of cerium oxalate precursor(a), and sample annealed at 200 ℃(b) and 400 ℃(c) in air
通过XRD谱图定性确认了热分解过程各个阶段的产物分别为Ce2(C2O4)3·9H2O、Ce2(C2O4)3和CeO2,结晶水的数目可以进一步由实验失重互相印证。
分析图2的热分析曲线,在100~180 ℃区间,DTG曲线出现第一个失重峰,DSC曲线表明该反应是一个吸热反应,发生结晶水脱去反应;180~290 ℃区间,TG曲线接近一个平台,没有化学反应发生;290~380 ℃区间,DTG曲线出现第二个失重峰,发生氧化热分解反应,DSC曲线表明该反应为放热反应,由于实验没有气体检测计量装置,结合文献可知无水草酸铈分解时放出CO和CO2[3,8],在空气之中,CO会与空气中的O2进一步反应生成CO2;380~700 ℃区间,TG曲线接近一个平台,失重反应停止,化学变化结束,但DSC曲线一直下降,表明最终样品,即CeO2随温度的升高热容不断变大,与空白坩埚对比产生吸热。
其中520 ℃处TG曲线有一个阶跃,DTG曲线出现一个增重峰,这可能是由于290~380 ℃时热分解步骤中部分草酸铈缺氧分解产生了Ce2O3,反应式为:

(1)
生成的Ce2O3在520 ℃与氧气反应生成最终产物CeO2,DSC曲线表明该反应是一个吸热反应,反应式为:
(2)
整个热分析曲线中,DSC曲线在TG曲线的平台阶段也是保持下降趋势,反应物质的总热容随着温度升高而不断变大。所得结果与Gallagher等[21]于1985年所得到的十三水草酸铈的热重历程基本一致。
根据热重数据可以得到草酸铈的理论失重和实验失重的对比列于表1。

表1 草酸铈在流动空气(20 mL/min)气氛中以10 ℃/min速率升温热分解的理论失重和实验失重对比Table 1 Comparison between experimental and theoretical mass losses for calcination of cerium oxalate at 10 ℃/min in streaming air(20 mL/min)
胡艳宏等[22]在开展的草酸铈前驱体热分解研究中,也采用草酸沉淀法自制草酸铈沉淀颗粒,得到的TG曲线反映样品的反应步骤1失重为21.937%,反应步骤2失重为30.276%;吕佳娉等[8]的草酸铈热分解研究中,自制草酸铈沉淀颗粒的反应步骤1的失重为21.9%。两者失重数据与本工作的实验失重数据相近,但小于反应步骤1的理论失重。这是因为实际加热时,处于中心的结晶水较难失去的缘故,上述两项工作也出现了中心结晶水脱去的延迟现象。反应步骤2的实验失重大于理论失重,而且理论剩余质量分数为48.76%,而实际最终剩余质量分数为47.13%,这可能有两个原因:一是反应步骤1中难以脱去的结晶水在这一步脱去,二是样品可能干燥程度不够,有部分的非结晶自由水一开始就存在于样品之中,这可以从100 ℃之前TG平台就已经出现失重加以印证。
综上所述,Ce2(C2O4)3·9H2O在空气气氛中的热分解机理如下:
100~180 ℃区间,发生结晶水脱去的吸热反应,反应式为:
(3)
290~380 ℃区间,发生氧化热分解的放热反应,总反应式为:
(4)
2.2 氩气气氛下U(C2O4)2·2H2O热分解机理研究
利用热重分析仪考察了U(C2O4)2·2H2O样品在氩气气氛中以10 ℃/min速率升温的热分解过程,热分析曲线示于图4。
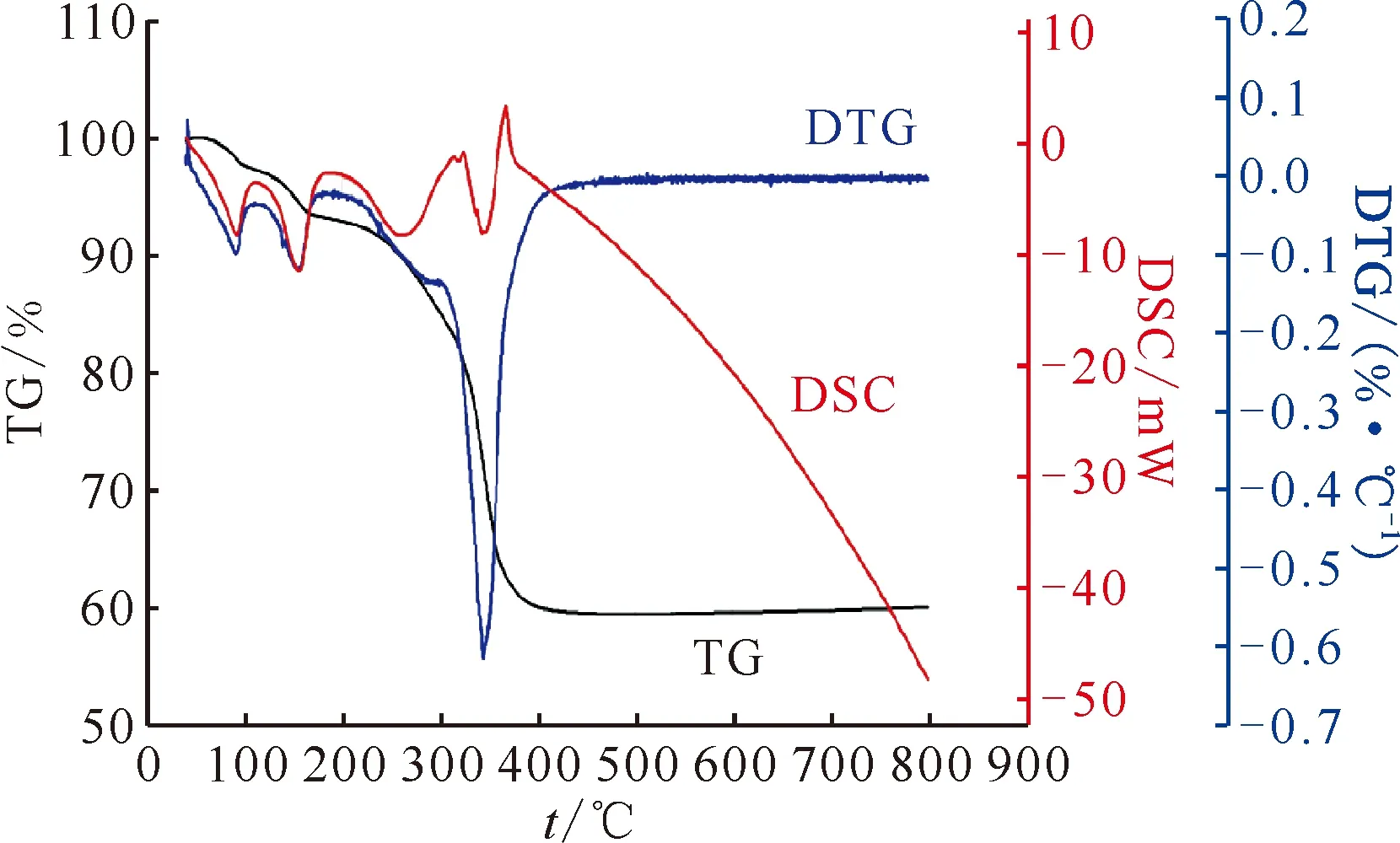
图4 U(C2O4)2·2H2O在氩气中以10 ℃/min速率升温的热分解过程Fig.4 Thermal decomposition of U(C2O4)2·2H2O on heating at 10 ℃/min in argon
利用管式炉获得氩气气氛下U(C2O4)2·2H2O 800 ℃退火反应产物,并将其与草酸铀前驱体一同进行XRD表征,并分别与U(C2O4)2·2H2O的标准卡片PDF#22-0961、UO2的标准卡片PDF#41-1422对比,结果示于图5。
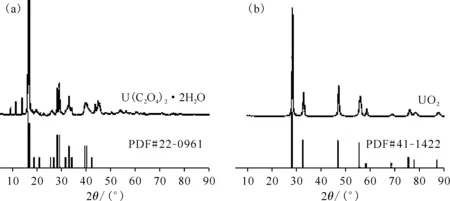
图5 氩气气氛下草酸铀前驱体(a)与800 ℃退火反应产物(b)的XRD图Fig.5 XRD spectrum of uranium oxalate precursor(a) and sample annealed at 800 ℃(b) in argon
草酸铀前驱体样品为绿色的粉末,结合热重曲线的脱结晶水质量百分比可以印证草酸铀结晶水的数目为2。800 ℃退火的反应产物为黑色的粉末,根据XRD图可以确定其为UO2。
分析U(C2O4)2·2H2O在氩气气氛下的热分析曲线,样品在68 ℃之前几乎无失重;68~140 ℃区间,DTG曲线出现第一个失重峰,脱去一分子结晶水,DSC曲线表明该反应为吸热反应,这与Ce2(C2O4)3·9H2O不同,由于其最后一个结晶水结合得比较紧密,它不是所有结晶水一起脱除,而是分步脱除,Wendlandt[23]观察到在脱去1个结晶水得到U(C2O4)2·H2O后热重曲线上出现了明显的平台;140~230 ℃区间,DTG曲线接着出现第二个失重峰,脱去最后一分子结晶水,DSC曲线表明该反应为吸热反应;230~400 ℃区间,DTG曲线出现第三个失重峰,发生分解反应,DTG曲线失重峰并不平滑,DSC曲线也表明该分解反应不是单纯的吸热或者放热反应,应该是多个子反应的复合反应,从Çlg等[10]关于草酸铀热分解的工作中可以知道,该过程包括草酸铀分解产生中间产物UO1.5(CO3)0.5以及中间产物的继续分解,还包括U2O5与UO2的相互转化;400 ℃以后直至800 ℃,TG曲线接近一个平台,没有进一步的失重反应,但DSC曲线一直下降,表明样品随温度的升高热容不断变大,与空白组对比产生吸热,可能产生相变。
氩气气氛下得到的草酸铀理论失重和实验失重的对比列于表2。

表2 草酸铀在氩气中以10 ℃/min速率升温热分解的理论失重和实验失重对比Table 2 Comparison between experimental and theoretical mass losses for calcination of uranium oxalate under at 10 ℃/min in argon
表2反应步骤1的实验失重略低于理论失重可能是由于中心结晶水脱除困难的原因,结晶水在后续的过程中被逐渐脱去,这就导致反应步骤3中的实验失重大于理论失重。由于最终产物为UO2,理论剩余质量分数为60.00%,而实验剩余质量分数为60.22%,与理论值非常吻合。
综上所述,U(C2O4)2·2H2O在氩气气氛下的热分解机理如下:
68~140 ℃区间,脱去第一分子结晶水,吸热反应,反应式为:
(5)
140~230 ℃区间,脱去最后一分子结晶水,吸热反应,反应式为:
(6)
230~400 ℃区间,无水草酸铀的热分解反应,无氧化还原,其总反应式为:
(7)
2.3 空气气氛下U(C2O4)2·2H2O热分解机理研究
利用热重分析仪考察了U(C2O4)2·2H2O样品在流动空气(20 mL/min)气氛中以10 ℃/min速率升温的热分解过程,热分析曲线示于图6。

图6 U(C2O4)2·2H2O在流动空气(20 mL/min)中以10 ℃/min速率升温的热分解过程Fig.6 Thermal decomposition of U(C2O4)2·2H2O on heating at 10 ℃/min in streaming air(20 mL/min)
利用马弗炉获得静态空气气氛下U(C2O4)2·2H2O在450 ℃和800 ℃下的退火反应产物,并将其与草酸铀前驱体一同进行XRD表征,并分别与U(C2O4)2·2H2O的标准卡片PDF#22-0961、UO3的标准卡片PDF#31-1416、U3O8的标准卡片PDF#47-1493对比,结果示于图7。此处马弗炉的气氛为静态空气,但对于流动空气气氛仍具有一定的参考性。
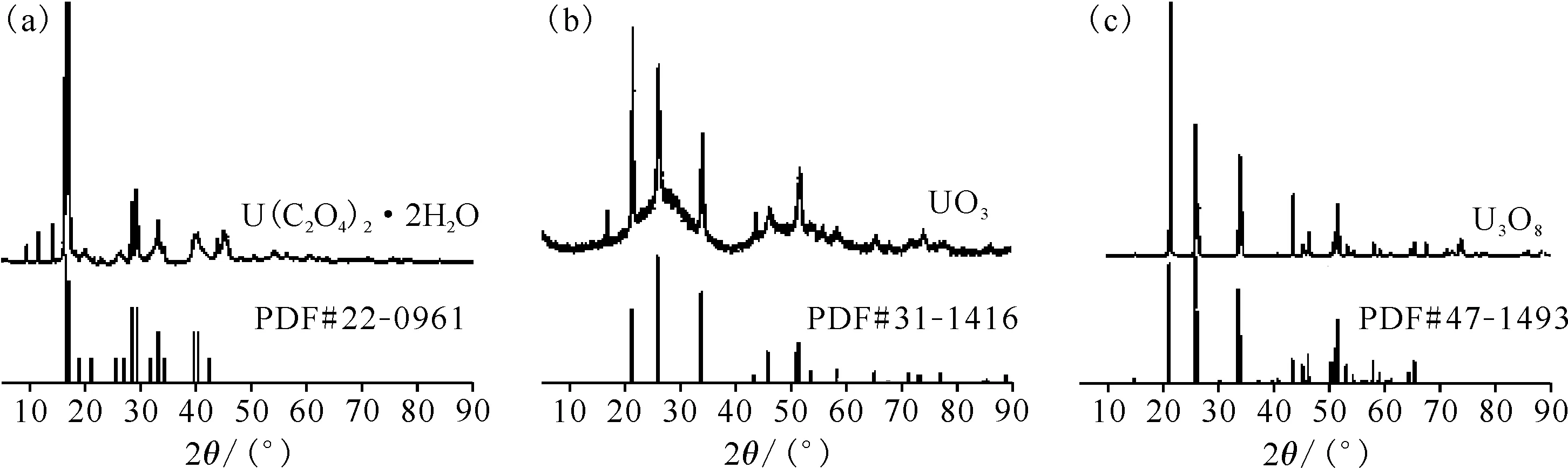
图7 草酸铀前驱体(a)和静态空气中450 ℃(b)、800 ℃(c)退火反应产物的XRD图Fig.7 XRD spectrum of uranium oxalate precursor(a), and sample annealed at 450 ℃(b) and 800 ℃(c) in static air
450 ℃退火反应产物粉末由主体橙红色颗粒和少量黑色颗粒构成(图8)。
XRD的结果与UO3的标准卡片PDF#31-1416吻合得很好,但同时与U3O8的标准卡片PDF#31-1425也有较好的吻合(图9)。
UO3化合物的颜色为橙红色,可以判断,用马弗炉在静态空气气氛下煅烧草酸铀,退火温度为450 ℃时产物为U3O8和UO3的混合物,对应热重曲线上的450 ℃热重平台的产物应当为UO3,这也与Çlg等[10]的结论保持一致。最终产物由XRD图可以确定是U3O8。

图8 静态空气中450 ℃退火反应产物Fig.8 Photo of sample annealed at 450 ℃ in static air

图9 静态空气中U(C2O4)2·2H2O 450 ℃退火反应产物XRD图(对比U3O8标准卡片)Fig.9 XRD spectrum of U(C2O4)2·2H2O sample annealed at 450 ℃ in static air(compared with U3O8 standard card)
分析U(C2O4)2·2H2O在空气气氛下的热分析曲线,样品在58 ℃之前几乎无失重;58~140 ℃区间,DTG曲线出现第一个失重峰,脱去一分子结晶水,DSC曲线表明该反应为吸热反应;140~240 ℃区间,DTG曲线紧接着出现第二个失重峰,脱去最后一分子结晶水,DSC曲线表明该反应为吸热反应;240~350 ℃区间,DTG曲线出现第三个失重峰,发生氧化分解反应,DSC曲线表明该分解反应是放热反应,DTG曲线失重峰并不平滑表明氧化分解也是一个复合的反应,从Çlg等[10]关于草酸铀热分解的工作中可以知道该过程包括草酸铀分解产生中间产物UO(CO3)、UO1.5(CO3)0.5以及中间产物的继续分解,还包括CO与氧气反应转化为CO2的过程;350~550 ℃,TG曲线接近一个平台,DTG无失重峰,DSC持续走低显示该阶段的产物随温度上升热容继续增大,与空白组对比产生吸热;550~600 ℃,DTG曲线出现一个较小的失重峰,发生氧化物转变过程,生成最终产物U3O8,从DSC曲线的斜率绝对值增大可以得知该反应为吸热反应。
空气气氛下得到的草酸铀理论失重和实验失重的对比列于表3。
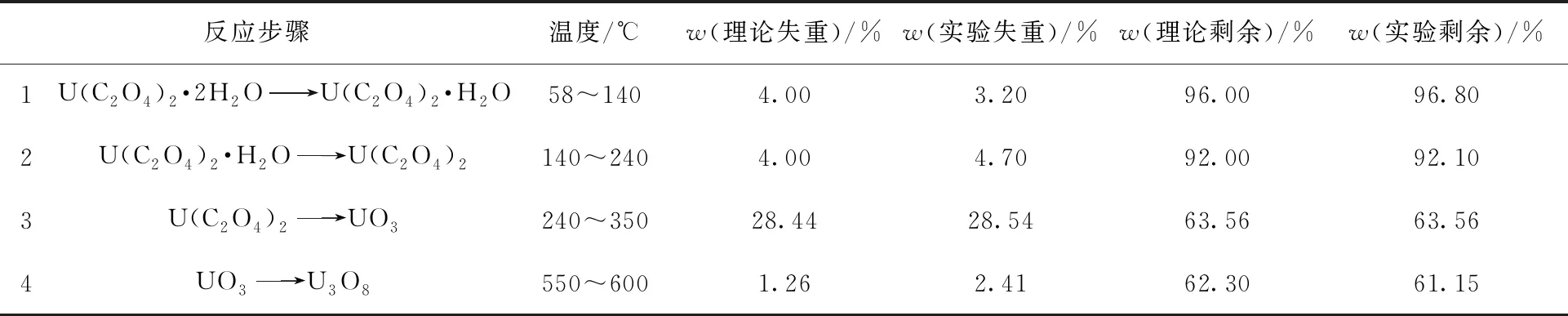
表3 草酸铀在流动空气(20 mL/min)中以10 ℃/min速率升温热分解的理论失重和实验失重对比Table 3 Comparison between experimental and theoretical mass losses for calcination of uranium oxalate at 10 ℃/min in streaming air(20 mL/min)
由表3可知,U(C2O4)2·2H2O在空气中失去结晶水的过程与在氩气中的类似,由于其最后一个结晶水结合得比较紧密,因而结晶水也是分步脱除,温度范围也相近,其反应步骤1的实验失重低于理论失重可能是由于中心结晶水脱除困难,但是在空气中后续的结晶水脱去过程明显更快,这就导致在反应步骤2中的实验失重大于理论失重,综合反应步骤1和反应步骤2的总体理论失重为8.00%,而实验失重为7.90%,总体上是相对吻合的。反应步骤3的理论失重与实验失重数据相差也不大,综合看前三个反应步骤,理论剩余质量分数为63.56%,实际剩余质量分数也为63.56%,吻合度较高。但是最后一个氧化物转换阶段理论和实验失重之间的差距较大,得不到较为合理的解释。在Çlg等[10]的研究中,U(C2O4)2·6H2O在空气中的最后氧化物转化过程的平均温度区间为838~1 000 K,即大致565~727 ℃的范围,失重平均为2.589%,转换成相对于U(C2O4)2·2H2O的失重为3.003%,仍然和理论失重有差别,因此氧化物转换步骤无法完全解释失重,有待进一步研究。
综上所述,U(C2O4)2·2H2O在空气气氛中的热分解机理为:
58~140 ℃区间,脱去第一分子结晶水,该反应为吸热反应,反应式为:
(8)
140~240 ℃区间,脱去最后一分子结晶水,该反应为吸热反应,反应式为:
(9)
240~350 ℃区间,发生复合的氧化分解反应,总反应为吸热反应,总反应式为:
(10)
550~600 ℃,发生氧化物转变过程,该反应为吸热反应,反应式为:
(11)
2.4 草酸铈、草酸铀煅烧前后形貌尺寸遗传性
表4是四组草酸铈前驱体以及它们在空气气氛下设置退火温度分别为400 ℃和950 ℃煅烧后产物的D50,其中D50是样品的累计粒度分布百分数达到50%时所对应的粒径,也叫中位径或中值粒径。本工作采用D50代表样品颗粒群的粒度大小。
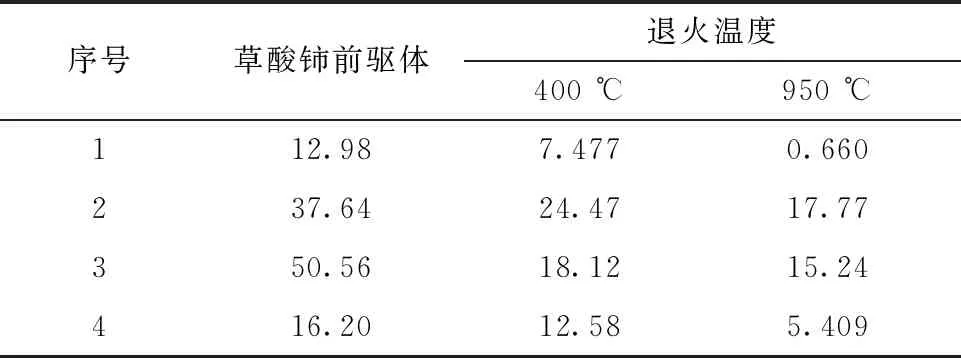
表4 草酸铈前驱体及空气气氛下400、950 ℃退火后煅烧产物的D50Table 4 D50 of cerium oxalate precursors and samples annealed at 400 ℃ and 950 ℃ in air μm
粒度分布直方图由于横坐标为对数坐标,难以直观看出粒度分布的宽窄,通过分别计算各个样品的粒度分布系数来表征样品粒度分布的宽窄,粒度分布系数数值越大代表分布越宽,并且一定程度上反映了团聚程度。其中粒度分布系数=(D90-D10)/2D50[24],其计算结果列于表5。
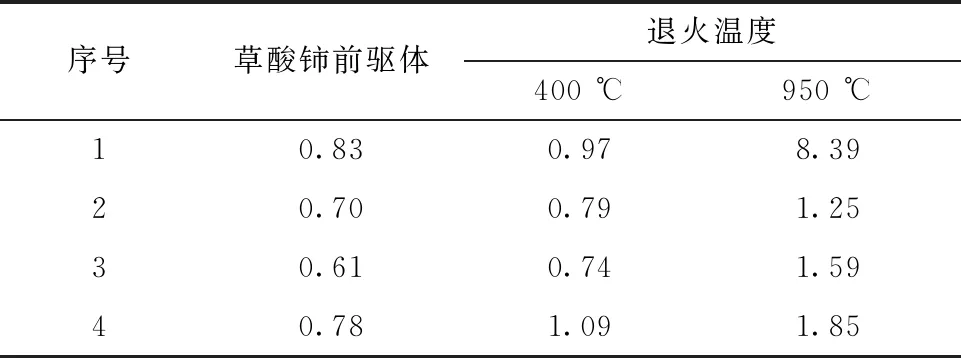
表5 草酸铈前驱体及空气气氛下400、950 ℃退火后煅烧产物的粒度分布系数Table 5 Particle size distribution coefficient of cerium oxalate precursors and samples annealed at 400 ℃ and 950 ℃ in air
表6是五组草酸铀前驱体以及它们在不同气氛不同退火温度下煅烧后产物的D50。

表6 草酸铀前驱体及不同气氛不同退火温度下煅烧产物的D50Table 6 D50 of uranium oxalate precursors and samples annealed at different temperatures in different atmosphere μm
其粒度分布系数计算后列于表7。
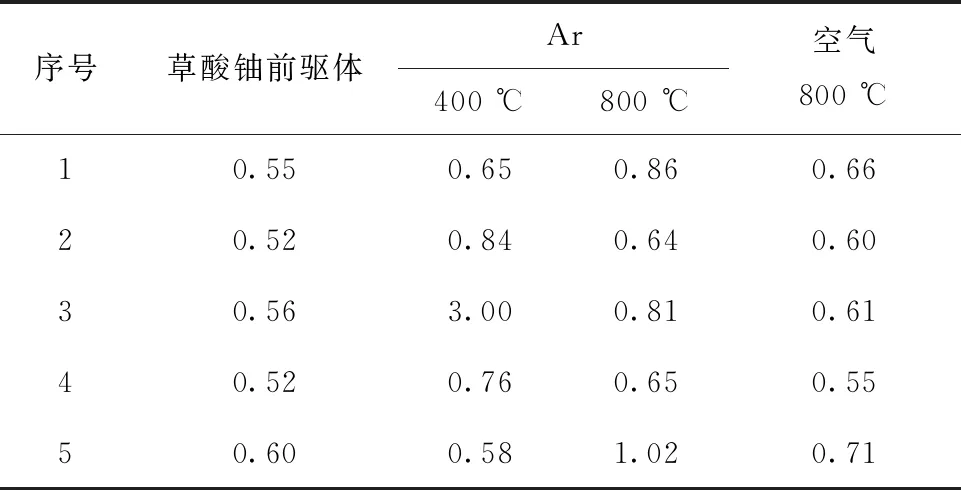
表7 草酸铀前驱体及不同气氛不同退火温度下煅烧产物的粒度分布系数Table 7 Particle size distribution coefficient of uranium oxalate precursors and samples annealed at different temperatures in different atmosphere
草酸盐煅烧前后粒度分布以第三组草酸铈样品的粒度分布图为例,如图10所示。
无论是草酸铈还是草酸铀,煅烧对粒度均造成两方面的影响。
1) 造成粒度一定程度的减小。从图10可以看到,退火产物的粒度图相比于前驱体的粒度图整体产生了左移,从表4和表6也可以看到,煅烧后退火产物的粒度相较于前驱体的粒度大多有明显的降低。马莹等[18]煅烧制备的氧化钇粒度相比其草酸钇前驱体也有一定程度上的减小。其中,表6的第5组数据在800 ℃煅烧之后粒度上升,这可能是由于颗粒烧结造成的。

(a)——前驱体,(b)——400 ℃,(c)——950 ℃图10 第3号草酸铈前驱体及其空气气氛下400、950 ℃退火后煅烧产物的粒径分布图Fig.10 Particle size distribution of the No.3 cerium oxalate precursors and samples annealed at 400 ℃ and 950 ℃ in air
2) 改变粒度分布,一般使粒度分布变宽。从图10来看,退火产物粒度图相比前驱体的粒度图变得更“胖”,即向低粒度和高粒度两方面延伸,粒度分布变宽,在图10(c)中,低粒度区甚至出现了小峰。从表5可以发现,草酸铈煅烧之后的退火产物粒度分布系数全都大于前驱体的,且随着退火温度的上升,其粒度分布系数也随之增大。从表7的粒度分布系数来看,大部分草酸铀样品煅烧之后粒度分布也变宽了。张亚文等[14-16]的研究发现短时间的灼烧能够有利于其颗粒分散,但长时间的灼烧下,颗粒会因为高温烧结形成严重的团聚体,因此粒度变宽可能是因为煅烧导致部分团聚消除产生低粒度颗粒,同时导致颗粒烧结团聚产生高粒度颗粒,两种效应导致颗粒的粒度分布发生改变,一般而言会导致粒度分布变宽。Ar气氛400 ℃下草酸铀第5号样品出现了粒度分布变窄,这可能是由于高温带来的烧结团聚的同时也导致极端低粒度的颗粒减小。
图11是Ce2(C2O4)3·9H2O 粉末和在静态空气气氛中分别经过400、950 ℃煅烧得到的氧化物粉末的SEM图。

图11 Ce2(C2O4)3·9H2O 粉末(a)和静态空气气氛中经400(b)、950 ℃(c)煅烧得到的氧化物粉末的SEM图Fig.11 SEM images of Ce2(C2O4)3·9H2O(a) and oxide powders obtained after calcination at 400 ℃(b) and 950 ℃(c) in static air
图12是U(C2O4)2·2H2O粉末和在不同气氛下分别经过400、800 ℃煅烧得到的氧化物粉末的SEM图。

(a)——U(C2O4)2·2H2O粉末,(b)——Ar气氛下400 ℃煅烧,(c)——Ar气氛下800 ℃煅烧,(d)——空气气氛下800 ℃煅烧图12 U(C2O4)2·2H2O粉末和在不同气氛中经400、800 ℃煅烧得到的氧化物粉末的SEM图Fig.12 SEM images of U(C2O4)2·2H2O and oxide powders obtained after calcination at 400 ℃ and 800 ℃ in different atmosphere
从图11可以看到,草酸铈前驱体以及煅烧产物主要为长条棒状,长度较短类似立方体,较长则为针状,并且经过煅烧虽然明显改变了团聚状态,但是形貌依然得以保留。从图12可以看到,草酸铀前驱体以及煅烧产物主要为方形片状,部分呈长条棒状,不同气氛和温度下的煅烧也基本保留前驱体形貌。这与Duvieubourg-Garela等[20]通过电镜观察U(C2O4)2·6H2O、U(C2O4)2·2H2O和U(C2O4)2·H2O晶体得到的结论一致。
3 结 论
针对草酸盐的热分解机理和煅烧前后颗粒形貌尺寸遗传性问题,本工作采用了同步热分析的方法,以XRD表征作为辅助手段,获取了草酸铈、草酸铀热分解过程的数据,阐释了热分解反应的机理。采用激光粒度仪和SEM对草酸盐前驱体煅烧前后尺寸形貌进行了表征,研究了煅烧过程前后尺寸形貌的遗传效应。主要结论如下:
(1) 空气气氛下Ce2(C2O4)3·9H2O的热分解反应主要分为两个阶段:第一个阶段失去所有结晶水,第二个阶段则是无水草酸铈氧化分解为CeO2;
(2) 氩气气氛下U(C2O4)2·2H2O的热分解反应也分为两个阶段:第一个阶段分步失去两分子结晶水,第二个阶段则是无水草酸铀分解为UO2;
(3) 空气气氛下U(C2O4)2·2H2O热分解反应则大体分为三个阶段:第一个阶段分步失去两分子结晶水,第二个阶段无水草酸铀发生氧化分解反应得到UO3,第三个阶段UO3发生氧化物转变反应,转变为U3O8;
(4) 对草酸铈和草酸铀的煅烧过程来说,煅烧会导致颗粒粒度一定程度减小,并会改变颗粒的粒度分布,一般是导致粒度分布变宽,但前驱体形貌能够基本保留,实验中草酸铈形貌主要为棒状,草酸铀的形貌主要为方形片状和棒状。
