基于TCGA数据库构建头颈部鳞癌RNA甲基化调控因子预后模型
2022-09-01彭颐宗丹葛宜枝何侠
彭颐, 宗丹, 葛宜枝, 何侠
头颈部鳞状细胞癌(head and neck squamous cell carcinoma,HNSCC)起源于口腔、咽部和喉部的黏膜上皮,是全球第6大常见的恶性肿瘤。全球每年估计有890 000例新病例和450 000例死亡病例[1]。手术、化疗、放疗和免疫治疗等多学科治疗方案能够显著提高HNSCC患者的生存时间和生活质量,但大多数患者会在3~5年内迅速进展,局部复发和远处转移的风险高达30%~60%,且复发和转移后存活率较低[2]。因此,早期识别复发转移高风险人群,并给予积极的临床干预有利于改善患者预后。
RNA甲基化是一种转录后水平调控基因表达的形式。其本质是基因片段上的碱基位点被甲基化后,参与调控RNA的翻译、剪切、表达与降解等生物学功能[3]。常见的RNA甲基化主要包括m6A、m5C、m1A与m7G等。RNA甲基化是一个可逆的过程,其修饰水平需依赖甲基转移酶、结合蛋白和去甲基化酶等的动态调节[4]。近年来的研究表明,RNA甲基化在肿瘤的发展与转归中发挥着重要作用,不仅能通过多种细胞信号通路驱动细胞恶性转化和化疗耐药等[4-5],且与患者的预后密切相关[6]。因此,利用在肿瘤中异常表达的RNA甲基化调控因子,探索RNA甲基化与肿瘤恶性进程之间的关联,对于基础研究或临床治疗肿瘤均有重要作用。
本研究旨在利用生物信息学寻找HNSCC组织中异常表达的RNA甲基化调控因子,探索这些基因对预测患者预后的价值,并建立相应的预后风险模型,为临床诊断及治疗提供新思路。
1 资料与方法
1.1 数据收集 从TCGA数据库(https://portal.gdc.cancer.gov/) 下载HNSCC患者肿瘤组织和正常头颈组织的转录组数据以及患者临床资料,包括年龄、性别、肿瘤分级与分期、生存时间与状态等。应用R统计软件(R version 4.1.1)中的Limma包对测序所得的基因表达谱做归一化处理。应用caret包,将HNSCC数据随机分成训练集与验证集用于后续的数据处理。
1.2 差异表达的RNA甲基化调控因子的鉴定 通过查阅最新文献,共选择了45种已知的m6A、m5C、m1A 和m7G的 RNA 甲基化调控因子[7-9]。利用R软件的Wilcoxon秩和检验鉴定在HNSCC样本与正常样本中差异表达的RNA甲基化调控因子(P<0.05,|log2 Fc|≥1),用pheatmap与igraph包绘制表达量热图与差异基因相关性网络图。通过 STRING(https://string-db.org)构建差异表达的RNA甲基化调控因子的蛋白相互作用(Protein-Protein Interaction Networks,PPI)网络。

1.4 预后预测模型的评估 为了评估模型的预测性能,对构建的模型进行Kaplan-Meier生存分析、受试者工作特征曲线(receiver operating characteristic,ROC)、PCA主成分分析与t-SNE降维分析,并采用单因素及多因素COX回归分析进行预后分析。利用高、低风险组间的差异表达基因,通过Cluster Profiler包进行GO富集分析。最后,进行了单样本基因集富集分析(ssGSEA),比较高、低风险组免疫细胞浸润差异。
2 结果
2.1 RNA甲基化调控因子差异表达分析 从TCGA数据库获得44例正常头颈组织以及502例HNSCC组织的转录组分析数据,并对45个RNA甲基化调控因子的表达水平进行比较,共鉴定出38个差异表达的基因(图1A)。其中,2个基因表达量在肿瘤组织中下降(YTHDC2、TET2),36个基因呈现出上调趋势。为进一步探索差异表达的RNA甲基化调控因子的相互作用,我们进行了蛋白质-蛋白质相互作用(PPI)网络分析(图1B)。设置PPI分析的最低交互评分为 0.7(高置信度),并确定了排名前10的中心基因(METTL3、ALKBH5、VIRMA、METTL14、WTAP、FTO、RBM15、YTHDC1、ALKBH1、ZC3H13)。此外,本研究还分析了RNA甲基化调控因子的表达相关性,相关性阈值取0.4(中等程度相关)(图1C)。
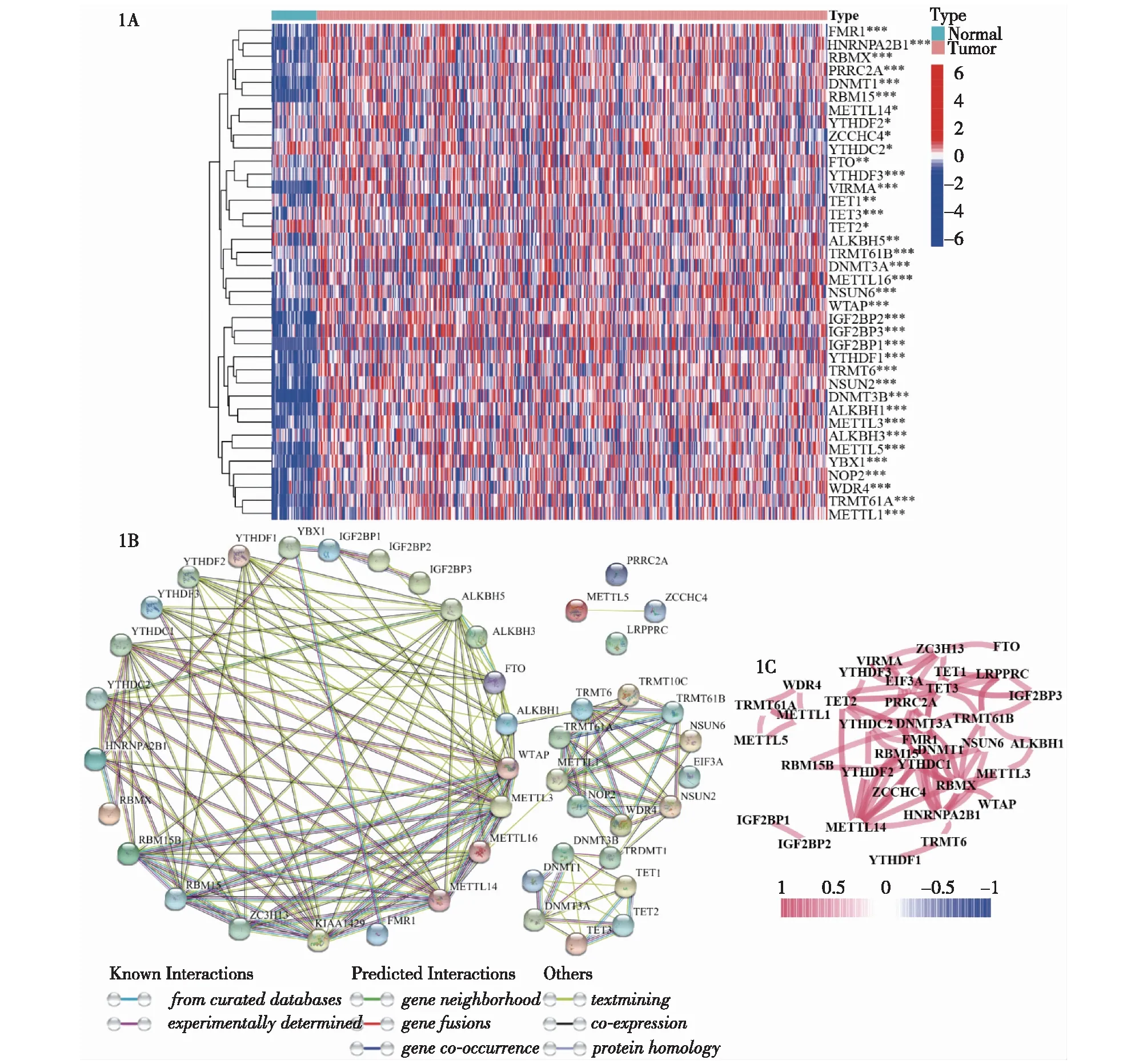
1A:正常组织和肿瘤组织之间 RNA 甲基化调控因子表达水平的热图(蓝色:低表达;红色:高表达;*P<0.05, **P<0.01,***P<0.001);1B:来自 STRING 的 RNA 甲基化调节因子的潜在相互作用;1C:RNA甲基化调控因子的共表达网络
2.2 头颈部鳞癌患者预后基因模型构建 将HNSCC患者数据分为训练集与验证集。在训练集中运用单因素COX回归分析初步筛选与生存相关的RNA甲基化调控因子(图2A),确定了图示的11个预后相关基因(P<0.2)。进一步对上述11个差异有统计学意义的RNA甲基化调控因子进行LASSO回归分析(图2B、2C)。根据λ值确定其中9个基因为模型的关键基因,并得出预后预测模型的风险评分公式如下:风险评分=[ExpYTHDC2×(-0.078)]+(ExpMETTL5×0.043)+(ExpHNRNPA2B1×0.016)+(ExpIGF2BP2×0.016)+(ExpIGF2BP1×0.031)+(ExpLRPPRC×0.005)+[ExpNSUN6×-(0.211)]+[ExpDNMT1×(-0.029)]+(ExpTRMT61B×0.107)。根据上述公式计算出的风险评分,将患者平均分为低风险组和高风险组(图2D)。主成分分析(PCA)和t-SNE分析显示,不同风险程度的两组患者分布模式存在一定差别(图2E、2F)。图2G显示了患者风险评分与生存状态的分布状态(图2G)。结果表明高风险组患者相对于低风险组死亡率更高,且生存时间更短。用Kaplan-Meier生存曲线评估该模型的预测能力,结果显示风险评分与HNSCC的生存相关,高风险组的5年生存率仅有43%,而低风险组的5年的生存率则相对较高为52%(P<0.001)(图2H)。时间依赖的受试者工作特征曲线(ROC)显示此风险模型在预测生存状态方面具有较好的诊断价值(1年AUC为0.619,3年AUC为0.653,5年AUC为0.601)(图2I)。
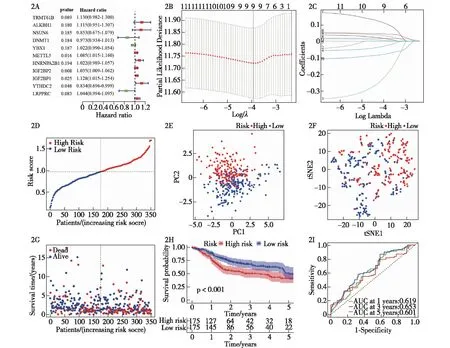
2A:与OS相关的RNA 甲基化调控因子;2B:LASSO 回归中参数选择的交叉验证;2C:LASSO 回归筛选变量动态过程图;2D:基于风险评分的患者分布情况;2E:主成分分析;2F:t-SNE分析;2G:每位患者的生存状态(低危人群:虚线左侧;高危人群:虚线右侧);2H:高风险组和低风险组患者 OS 的Kaplan-Meier 曲线;2I 预后模型的ROC曲线
2.3 预后模型的验证 根据训练集建立的预后模型风险评分公式计算验证集中病例的风险值,取中位风险评分,将验证集分为高风险组与低风险组(图3A)。主成分分析(PCA)和t-SNE分析显示高、低风险组具有不同的分布模式(图3B、3C)。低风险组的患者比高风险亚组中的患者具有更长的生存时间和更低的死亡率(图3D)。Kaplan-Meier生存曲线还证明低风险组的存活率明显高于高风险组的存活率(P<0.001)(图3E)。ROC曲线分析显示此模型有预测效力(1年AUC为0.614,3年AUC为0.649,5年AUC为0.673)(图3F)。
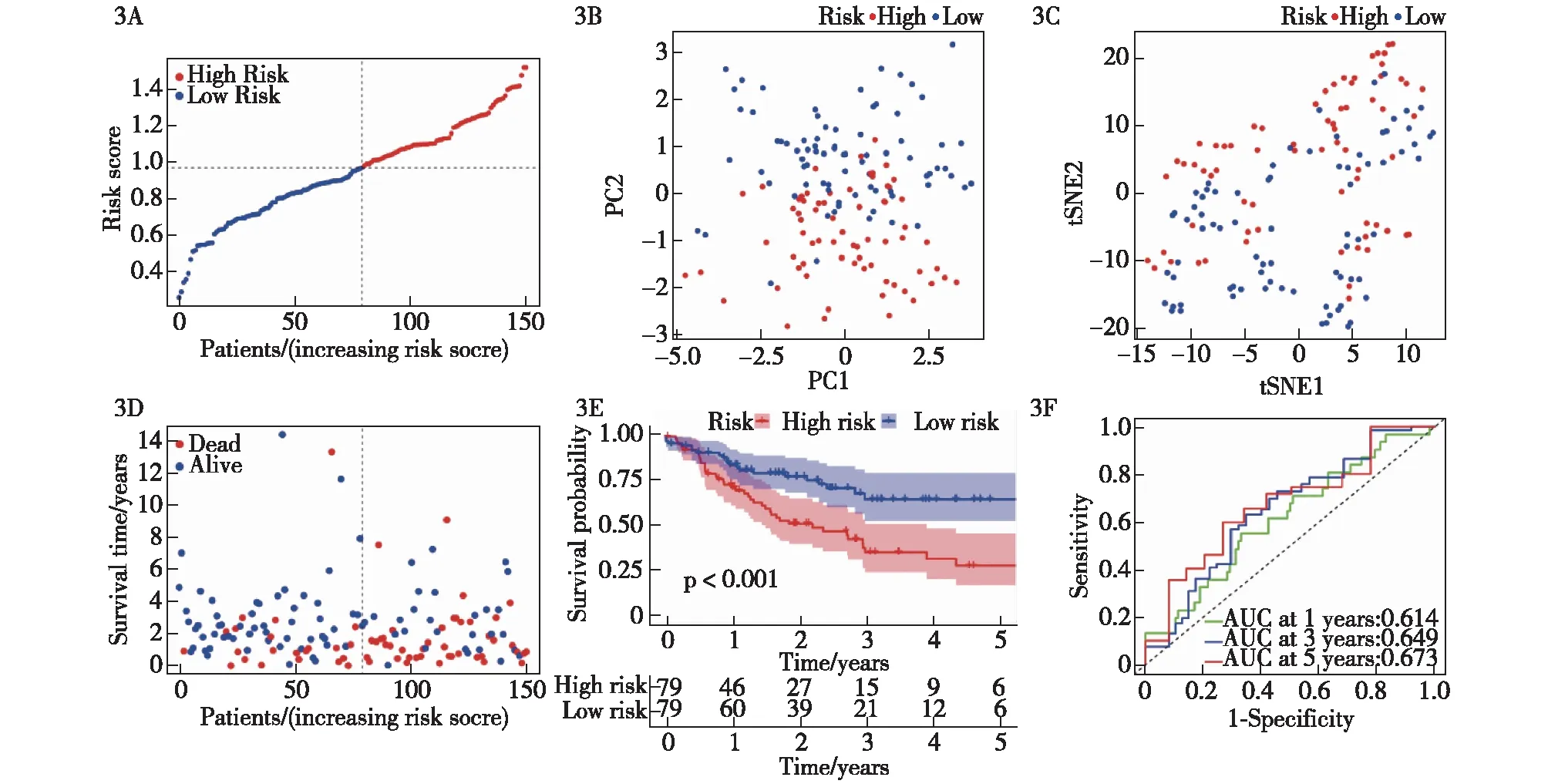
3A:基于风险评分的患者分布情况;3B:主成分分析;3C: t-SNE分析;3D:每位患者的生存状态(低危人群:虚线左侧;高危人群:虚线右侧);3E:高风险组和低风险组患者 OS 的Kaplan-Meier 曲线;3F:预后模型的ROC曲线
2.4 风险模式的独立预后价值 单因素和多因素COX回归分析来评估RNA甲基化调控因子预后预测模型。单因素COX回归分析表明,该模型的风险评分是预测TCGA队列预后因素(HR=3.791,95%CI:1.477~9.726)(图4A)。多因素分析同样表明,在调整其他混杂因素后,风险评分能够作为HNSCC患者的独立预后因素(HR=3.436,95%CI:1.299~9.093) (图4B)。此外,患者的T分期、N分期与M分期情况同样也可作为独立的预后因素。本研究还做了高、低风险组患者的临床特征与预后预测模型中9个关键基因表达量的热图(图4C)。结果显示高、低风险组的患者在肿瘤分期与T分期方面,差异有统计学意义(P<0.01)。
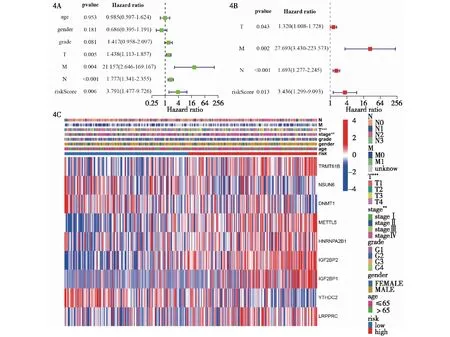
4A:TCGA 队列风险评分的单因素COX回归分析;4B:TCGA 队列风险评分的多因素COX回归分析;4C:临床病理特征和风险组之间联系的热图(蓝色:低表达;红色:高表达;**P<0.01; ***P<0.001)
2.5 基于风险模型的富集分析 在TCGA数据中,提取风险模型分类中高、低风险组之间差异表达的基因共2 630个。其中,1 576个基因在高危组中高表达,1 054个基因在高危组中低表达。基于这些差异表达基因进行GO富集分析。结果显示,差异表达的基因主要参与到免疫应答激活信号转导、细胞吞噬作用以及免疫球蛋白相关的免疫应答等机体免疫相关的生物学过程(图5)。
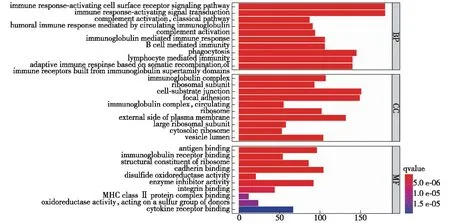
注:条形越长表示富集的基因越多,红色越深表示差异越明显;q值表示调整后的P值
2.6 基于风险模型的免疫功能比较 运用单样本基因集富集分析(ssGSEA),比较了HNSCC高、低风险组免疫细胞的浸润水平与免疫相关通路的差异。在TCGA数据中,高风险组的免疫细胞浸润水平普遍低于低风险组(图6A)。除巨噬细胞(Macrophages)浸润水平降低不具有显著性外,其余13种免疫细胞浸润水平,差异有统计学意义(P<0.001)。免疫相关通路的差异分析也显示,高风险组相较于低风险组存在普遍的免疫相关通路的抑制现象,有11种免疫相关通路均受到了不同程度的抑制(P<0.05)(图6B)。

6A:低风险组(蓝框)和高风险组(红框)之间16种免疫细胞富集评分的比较;6B;低风险组与高风险组之间13种免疫相关通路类型富集评分的比较
3 讨论
近年来,RNA甲基化在mRNA、tRNA、rRNA、snRNA等各种类型的RNA中的作用被广泛关注,并日益成为肿瘤研究领域中的热点。RNA甲基化水平的调控受到3类蛋白的影响:负责写入甲基化的甲基转移酶,能够擦除甲基化的去甲基化酶,以及识别甲基化位点行使生物学功能的甲基化阅读蛋白。多项研究证实RNA甲基化修饰能够通过多种机制影响肿瘤的发生和发展,例如:参与肿瘤干细胞多功能性的形成与分化[10],参与肿瘤细胞增殖[11]以及参与肿瘤细胞侵袭转移[12]等,并且这些异常的RNA甲基化修饰的改变通常导致患者的不良预后[13]。此外,Li等[14]的研究证实,m6A甲基化修饰能够通过靶向IL-7/STAT5/SOCS通路调控T细胞的稳态,这意味着RNA甲基化在肿瘤免疫中也扮演着不可替代的角色。因此,本研究着重关注RNA甲基化修饰与肿瘤及肿瘤免疫的关系。
RNA甲基化是目前发现的170多种RNA转录后修饰中最主要的修饰方式之一,包括m6A、m5C、 m1A和m7G等。Chen等[15]的研究基于3个m6A调控因子(METTL4、METTL3、HNRNPC)建立了一个用于预测HNSCC预后的模型。该模型划分的高、低风险组能够很好地估计患者的生存状态,但是患者的风险评分与其他临床病理特征之间并没有显示出明显的相关性。类似的文章还见于Han等[16]的研究,该研究基于m5C调控因子(NSUN5、DNMT1、DNMT3A)建立了HNSCC预后预测模型。然而,最近的一项研究表明在不同癌症类型中,m6A与5mC调控因子之间存在表达的高度相关性,意味着m6A与5mC调控因子可能存在串扰现象。鉴于这种不同种类RNA甲基化修饰潜在的关联,本研究选择使用4种RNA甲基化的调控因子共同构建预后模型。其中,RNA m6A甲基化是RNA最常见的转录后修饰,在本模型中也占据主导地位。m5C、m1A的调控因子也都参与到模型的构建。而m7G的调控因子并未参与到风险模型的构建,考虑由于m7G甲基化的研究尚处于起步阶段,目前已鉴定的调控因子过少导致。
本研究的预后模型涉及9个RNA甲基化调控因子,包括4个甲基转移酶与5个结合蛋白。METTL5能够催化18S rRNA的m6A甲基化并促进肿瘤细胞生长与胚胎干细胞的分化[17-18]。TRMT61B则主要作为m1A甲基转移酶催化线粒体的16S rRNA与tRNA[19]。DNMT1与NSUN6能够催化m5C修饰,DNMT1已被证实在口咽癌、喉鳞癌等HNSCC过度表达,并且与不良预后相关[20-21]。NSUN6则是具有较强底物特异性的mRNA与tRNA m5C甲基转移酶[22-23]。与本研究不同的是,NSUN6在胰腺癌中被鉴定为表达量下调,并可以通过调节细胞增殖抑制胰腺癌的发展[24]。NSUN6在HNSCC中的潜在生物学作用及机制还需进一步研究来揭示。结合蛋白IGF2BPs家族在多项研究中被报道与mRNA的稳定性和翻译相关。IGF2BP1与IGF2BP2能够分别通过介导BMI1的翻译[25]与提高c-Myc的mRNA稳定性[26]促进口腔鳞癌的恶性进展。hnRNP家族是另一类m6A结合蛋白,有报道认为在HNSCC中HNRNPA2B1通过调节癌基因的剪接事件促进上皮-间充质转化[27]。而高甲基化的LRPPRC在舌鳞状细胞癌中被认为具有一定诊断价值和预后预测潜力[28]。模型中比较特殊的m6A结合蛋白YTHDC2是为数不多的在HNSCC中表达量下降的调控因子,已被证实是与HNSCC预后、凋亡激活和泛素介导的蛋白水解有关的抑癌基因[29-30]。因此,本模型涉及的RNA甲基化调控因子与肿瘤的恶性进展,尤其是HNSCC的增殖与侵袭存在着密不可分的关系,用此模型来预测HNSCC患者的预后是有据可循的。
有研究证据表明RNA甲基化修饰及其调控因子在病原体识别、免疫细胞激活和免疫反应中起重要作用[31]。是免疫系统稳态和激活的新型调节剂,也是肿瘤发生、转移、治疗抵抗和复发的切入点。本研究模型利用RNA甲基化调控因子表达量的差异划分的高、低风险组之间存在明显的免疫功能与免疫浸润差异,恰也证明了以上的观点。
综上所述,RNA甲基化调控因子在HNSCC中的异常表达可能在肿瘤的发生发展中起重要作用。本研究基于9个RNA甲基化相关调控因子构建的预后模型,能有效对HNSCC患者进行高、低风险的分层,具有良好的预后预测效力,并且通过了验证集的检验,能够为临床制定合理个体化治疗方案提供参考。然而,本研究结果仍需要大量临床数据的支持与多中心的循证医学证据的验证,RNA甲基化与机体免疫的作用相关性也有待进一步发掘。
