我国主要沿海地区O1群霍乱弧菌分子分型特征分析
2022-08-09柳婷婷杨文辉金大智王景林辛文文
柳婷婷,李 萍,杨文辉,金大智,高 姗,王 菁,王景林,康 琳,辛文文
霍乱弧菌(Vibriocholerae)属于革兰阴性菌,普遍存在于不同的水生环境中。根据其主要表面耐热抗原O,目前霍乱弧菌可分为近200多种血清群[1],其中O1群和O139群能够产生霍乱毒素(Cholera Toxin,CT),是引起霍乱的病原菌。O1血清群可进一步分为3种血清型——小川型(Ogawa)、稻叶型(Inaba)和彦岛型(Hikojima)[2]。
霍乱是人类感染了产毒素的霍乱弧菌而引起的急性水样腹泻病,暴发突然且易传播,死亡率高,属于国际检疫传染病[3]。病原菌分子分型是追溯霍乱传染源,调查传播途径的主要手段。霍乱弧菌有许多种血清型,不同血清型的细菌,生理生化特性可能相同,但是对人类和动物的致病性可能完全不同。因此,对霍乱弧菌进行快速分型具有重要意义。目前以核酸序列差异分析为主的分子分型手段对霍乱弧菌进行分型与进化的研究是较为先进且最为灵敏准确的,主要方法包括肠杆菌基因间重复一致序列(Enterobacterial Repetitive Intergenic Consensus, ERIC)PCR[4]、脉冲场凝胶电泳(Pulsed Field Gel Electrophoresis, PFGE)及多位点序列分型(Multilocus Sequence Typing, MLST)技术等[5]。MLST技术能够在无细菌纯培养物的情况下,直接从临床样本中扩增出特定位点基因片段进行分析,直观反映等位基因的变异情况,并且能够实现全球范围内的实验室数据交流共享。随着基因组测序成本的降低,目前正处于从MLST到全基因组测序分型的过渡阶段[6]。基质辅助激光解吸电离飞行时间质谱(Matrix-assisted Laser Desorption/Ionization-time of Flight Mass Spectrometry, MALDI-TOF MS)具有快速准确、灵敏度高、分辨率高等优点,已广泛应用于临床病原微生物的鉴定[7]。MALDI-TOF MS具有亚型区分的潜力,是研究菌株亲缘关系、确定暴发菌株和地域特征间密切关联的有力工具[8]。目前,MALDI-TOF MS对多血清型细菌的鉴定和分类仍处于起步阶段,如对副溶血弧菌O3群等均表现出较好的分型能力[9],在对艰难梭菌基因型ST37的表征区分[10]、流行性霍乱弧菌O1/O139群与非流行性霍乱弧菌[11]的快速区分也表现出了良好的效果,但是在其他物种的应用上可能会受限制,需要实验进行分析。
本研究利用MLST分型方法鉴定192株霍乱弧菌的MLST分型,进而分析其种群结构。此外,利用MALDI-TOF MS技术分析霍乱弧菌蛋白指纹图谱的相关性,并对两种方法的结果进行比较。部分MLST数据库中的O1群菌株也纳入研究体系中,以进行相关的比较分析。
1 材料与方法
1.1 实验材料 本实验采用的霍乱弧菌核酸和灭活蛋白来自863计划项目(课题编号:2014AA021402)参与单位浙江省疾病预防控制中心,其中大部分属于O1血清群,包括Inaba 63株、Ogawa 125株。其余4个菌株没有相关血清型信息。
1.2 PCR扩增 根据霍乱弧菌MLST数据库提供的方案,选择adk、gyrB、mdh、metE、pntA、purM和pyrC这7个管家基因作为靶基因进行扩增(表1)。PCR反应体系为50 μL,包括正向和反向引物各0.5 μL(30 pmol/mL)、0.5 μL 10 mmol/L dNTP、5μL 10× Buffer(500 mmol/L KCl、100 mmol/L Tris-HCl pH8.0、1%TritonH X-100及15 mmol/L MgCl2)、2U的ExTaq聚合酶(TaKaRa)和5 μL模板DNA,加ddH2O至总体积为50 μL。PCR的反应条件:95 ℃预变性5 min;94 ℃ 50 s、56 ℃ 50 s、72 ℃ 80 s,进行30个循环;72 ℃延伸7 min。使用PCR引物在ABI-3700测序仪上对扩增子进行双向测序,并使用Contig-Express进行比对。使用Bioedit和MUSCLE version 3.8等软件进行多序列比对和比较分析。对于16S rDNA,PCR扩增体系为25 μL,包含正向和反向引物各1 μL、12.5 μL的2×Taq PCR master Mix、2 μL模板DNA,加ddH2O至总体积为25 μL。PCR反应条件:94 ℃预变性5 min;94 ℃ 60 s、50 ℃ 60 s、72 ℃ 60 s,进行30个循环;72 ℃延伸5 min。

表1 霍乱弧菌基因组7个等位基因MLST扩增引物
1.3 MALDI-TOF MS分析的样品制备 首先使菌体蛋白(约10 mg)完全悬浮于300 μL ddH2O中,然后加入900 μL无水乙醇吹吸均匀,进行灭菌;12 000 r/min离心2 min后去除上清液(尽可能去除干净);向沉淀中加入50 μL 70%的甲酸,使菌体蛋白完全悬浮后再加入50 μL乙腈,充分混匀后12 000 r/min离心2 min;取1 μL上清液进行靶板点样,并在室温下干燥。样本干燥后立即向样本上覆盖2 μL的α-氰基-4-羟基肉桂酸(α-cyano-4-hydroxycinnamicacid, CHCA)并在室温下干燥形成共结晶,后续即可上机分析。
1.4 序列多样性分析 使用DnaSP 5.0计算7个等位基因的多样性指数π、G+C含量及单核苷酸多态性(Single Nucleotide Polymorphism, SNP)个数。使用KaKs Calculator Version 2.0计算非同义和同义突变的比值dN/dS。
1.5 等位基因多样性分析 将7个等位基因序列按照adk、gyrB、mdh、metE、pntA、purM、pyrC的顺序拼接在一起,将拼接序列结果与霍乱弧菌MLST数据库进行比对,确定其等位基因型(Allelic Profile, AP)和序列型(Sequence Type, ST)。新的AP和ST提交到PubMLST数据库。依据序列型的等位基因信息,使用eBURST Version 3.0对STs进行分析,归类为克隆群(Clonal Complex, CC)。CC包含至少3个基因位点不同的STs,7个位点中有6个及以上位点相同则定义为遗传相关克隆。
1.6 种群结构分析 利用STRUCTURE 2.2,采用马尔可夫链蒙特卡罗(Markov Chain Monte Carlo, MCMC)算法,分别分析评估本试验得到的22个STs和40个数据库中现有的O1/O139群相关STs。计算种群内群体数量K值,各个序列型间依据同源性高低而确定群体归属。本研究中,K值设置范围为2~10,每100 000次重复并弃去前20 000次不稳定结果。
1.7 系统发育树分析 使用MUSCLE 3.8.31比对O1血清群的22个STs和40个STs的连接序列,并通过MEGA 5.0对获得的核酸序列进行进化分析(1 000次重复),构建无根系统发育树。利用SplitsTree 4.0软件,采用Neighbour-net算法构建16S rDNA序列的网状结构图,检验种群间是否存在重组现象。
1.8 MALDI-TOF MS图谱生成和聚类分析 采用正线性模式,每种菌株采集4~5张复合图谱,每张复合图谱由6张单图谱组成。通过基线减法和平滑处理进行预处理。将属于同一ST的分离株的蛋白指纹图谱进行叠加,使用BioTyper 3.0软件(Bruker Daltonics)中的PCA算法进行蛋白指纹图谱分析,以便分析不同ST之间的差异。
2 结 果
2.1 核苷酸位点和等位基因序列多样性 7个等位基因的遗传多样性结果如表2所示。等位基因型最多的是metE,共11个,最少的是purM,有3个。位于II号染色体上的pntA和pyrC均有10个等位基因型。pyrC共含有84个SNPs,显示出最高的遗传多样性,其次是metE,含有58个SNPs。purM是最保守的基因,只有3个SNPs,且π是7个管家基因中最低的。pyrC的π水平最高,为0.060 58。除了pntA存在正选择效应外,其余等位基因均受到自然纯化作用。

表2 核苷酸和等位基因序列多样性
2.2 序列类型和克隆群 本研究22个STs中11个为新鉴定的ST。ST69为最主要的序列型,192株菌株中82株为ST69,其次是ST75,有40个菌株。新鉴定出的STs中,ST176包含的菌株数最多,有5个菌株。
图1A显示的是22个STs之间单等位基因位点变异关系(即7个等位基因中,至少有6个等位基因型相同)。22个STs形成2个CCs,均包含5个STs。CC176包含的5个STs分别是ST171、ST172、ST173、ST176和ST177,其中ST176为该CC的founder。CC430包含的5个STs分别是ST164、ST165、ST174、ST430和ST431,ST430为该CC的founder。剩下的12个STs形成了3个双体和6个单体,ST69与ST75被分到两个不同的双体中。在对霍乱弧菌MLST数据库中O1/O139血清群对应的40个STs进行分析时,ST69与ST75分别形成了CC。CC69包含ST69、ST70和ST429,CC75包含ST75、ST169、ST170和ST182(图1B)。图1C显示的是与数据库中非O1/O139血清群STs的单等位基因位点变异关系,本研究中22个STs以红色圆圈标出。结果显示,所有STs共形成6个CCs,分别为CC430、CC176、CC270、CC204、CC8和CC499。本研究中的22个STs未与数据库中非O1/O139群对应的STs形成新的克隆群。

注:彼此为单等位基因位点变异的ST用黑线连接。黑色圆点的大小与每个ST内的菌株数量有关,圆点越大数量越多。每个CC的名称用红色标出。
2.3 霍乱弧菌的系统发育和蛋白指纹图谱分析 使用MEGA 5.0构建邻接(Neighbor joining, NJ)树(图2A)以分析本研究中22个STs与MLST数据库中O1/O139群对应的40个STs之间的系统发育关系。22个STs形成2个谱系,ST169、ST75、ST429和ST69聚集在一个谱系中,其他18个STs在另一个谱系中。数据库中40个STs之间的关系也分为2个谱系,2个谱系中都包含本试验的22个STs(数据未显示)。
采用neighbour-net方法,根据16S rDNA序列构建了各个STs间分支网状图,如图2B所示。192个菌株共含有13种不同的16S rDNA序列,其中8种序列包含1株以上的分离株和ST型。13种不同的序列中,4种序列都包含ST173,说明ST173分离株对应的16S rDNA序列具有多样性。此外,不同的16S rDNA序列存在共有的ST,如ST69、ST75和ST173。
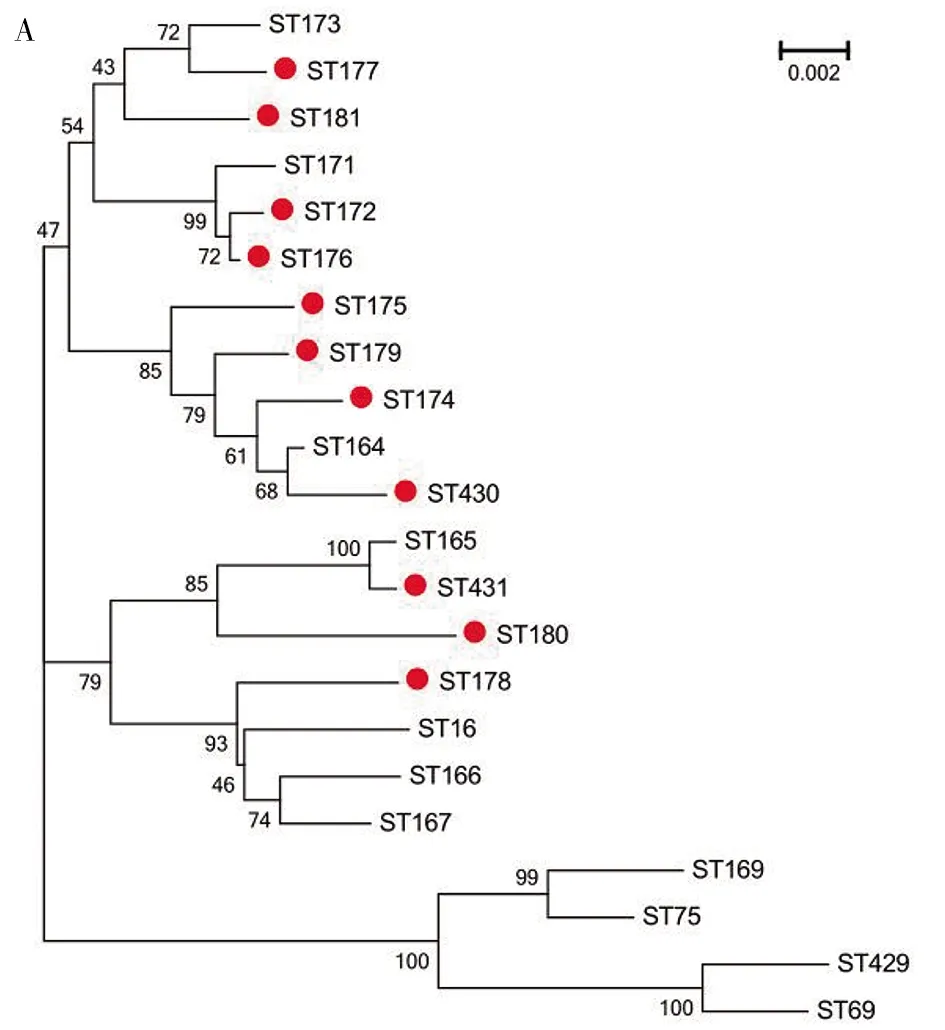
A:采用最大似然法计算22个STs间的进化距离,构建系统发育树,新的ST用红色圆点标出;B:采用neighbour-net算法计算192株霍乱弧菌的16S rDNA序列与22个STs间的关系,并绘制成分支网状图,ST后括号内的数字表示属于同一ST的分离株数。
图3A显示了分别属于22个STs的分离株的蛋白指纹图谱的胶图。不同STs的图谱基本相同,质量峰(m/z)均在2 000~12 500 Da范围内。另外,采用BioTyper 3.0对本研究中的22个STs的蛋白指纹图谱的相关性也进行了分析,结果如图3B所示。当距离为200时,22个STs被分为2组,较大的组包含除ST171外的21个STs。当距离为100时,22个STs被分成3组,ST179又形成了一个单独的组。蛋白指纹图谱分析结果与核苷酸序列分析结果不存在明显相关性。

A:22个STs的蛋白指纹图谱胶图。相同ST的分离株图谱叠加,视为1株分离株;B:根据22个STs的蛋白指纹谱生成的聚类分析树状图,属于相同ST的分离株视为1株分离株。
2.4 种群结构 利用LDhat评估了霍乱弧菌7个等位基因的重组率(rho)与突变率(theta),每个基因的重组率(purM除外)如表3所示。它们的比率范围从0.31(gyrB)到388.52(adk)不等,7条序列总体比率为4.24。对于本研究中22个STs而言,在进化过程中,重组比突变更容易发生。此外,对霍乱弧菌MLST数据库中已提交的属于O1/O139血清群的40个STs,及所有霍乱弧菌的364个STs分析发现,包含所有STs的组的rho高于致病血清群STs的rho,平均重组率为863.46,而另外两组的重组率均<1。IA值均>0,表明在a、b、c 3个组中均存在连锁不平衡的倾向(表4)。

表3 各基因的重组检测和评估
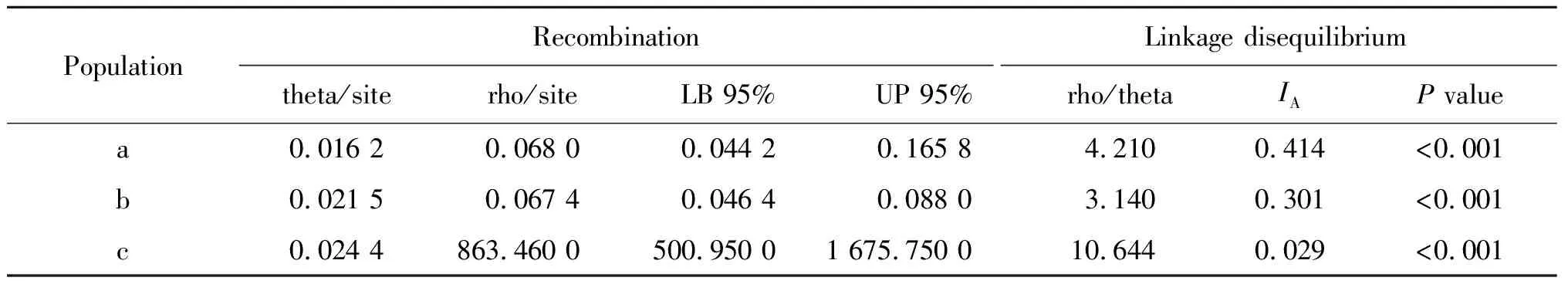
表4 各种群基因的重组检测和评估
采用STRUCTURE 2.2进一步分析种群结构,发现22个STs被分成3个亚群,即K=3(图4A)。ST69、ST429、ST75和ST169主要存在于第I组(红色组)中,与第II组(绿色组)和第III组(黄色组)存在少量交叉。第II组包含11个STs,其中8个STs是本研究中新发现的,第III组中包含3个新发现的STs。图4A中第I组(红色组)包含的4个STs在图4B中同样也分布在同一组(黄色组)。图4A中其余18个STs在图4B中分布在同一个组(红色组)。值得注意的是,图4B中黄色组位于红色组的中间,并将红色组分为2个部分,表明黄色组在红色组别间形成了“亚克隆群”。ST189、ST191和ST184存在2个不同组别间(绿色组和红色组)的重组现象。为进一步分析O1群对应的STs和非O1/O139群对应的STs间的种群结构关系。我们也对数据库中的STs进行了种群结构分析(图4C)。结果显示,所有的STs共形成4个亚群,绝大部分的STs都存在于红色组,紫色和绿色对应的亚群穿插在红色组中,黄色组被红色组分为距离较远的2个部分。本研究中的22个STs全部存在于红色组。

A:本研究中22个STs的种群结构图;B:数据库中40个O1/O139血清群对应的STs构建的种群结构图;C:数据库中STs构建的种群结构图。不同的颜色代表不同的种群。
3 讨 论
霍乱弧菌是霍乱的病原菌,通常会引起成人和儿童急性腹泻疾病,是我国甲类肠道传染病。目前已经有许多针对霍乱弧菌O1血清群分离株的研究[12],本研究选择MLST方法来评估192株O1群霍乱弧菌的遗传多样性。MLST计算的是管家基因核酸位点的变化,不受操作条件的干扰,可实现最大的数据共享。霍乱弧菌已建立了MLST数据库(www.pubmlst.org/vcholerae),并且已有很多学者利用该方法对霍乱弧菌进行研究。此外,MALDI-TOF MS可以作为一种潜在的亚型区分方法区分大肠杆菌O157、O26和O111与其他血清型[13],不过在某些物种的应用上也会受限制[14]。
本研究中的菌株大多属于血清型Inaba或Ogawa,Ogawa是最普遍的血清型。O1群的另一种血清型Hikojima较罕见且不稳定。本研究发现最保守的基因是purM,变化最大的基因是pyrC,这一结果与其他菌株的研究一致。此外,我们还发现7个管家基因在进化过程中,重组比突变更容易发生,尤其是adk基因的比率为388.52,远高于其他研究中非O1/O139菌株的比率[12]。霍乱弧菌非O1/O139群与O1/O139群在系统发育上关系较远[15]。类似研究结果也表明,非O1/O139群霍乱弧菌对应的STs常形成单体,种群内部存在异质性[16]。本研究中,O1群对应的STs与数据库中非O1/O139群对应的STs未能形成新的克隆群,且绝大部分非O1/O139血清群对应的STs如星星状散在分布,与上述研究结果相似。
ST69是第7次霍乱大流行分离株中的主要ST。类似的研究结果也表明,绝大部分O1/O139临床菌株都是ST69[17],但含有不同的毒力基因性状,这可能是基因水平转移导致[18]。近年来,研究发现我国多地主导的ST 69被ST75所取代[19-20]。非O1/O139菌株比O1/O139菌株表现出更高的遗传多样性,可能是由基因重组和/或突变引起[12]。ST69印度毒株与全球毒株之间的系统发育学分析表明,ST69与ST429密切相关[21],ST429由ST71突变而来。本研究的16S rDNA序列的Neighbor-net网状分析结果表明,不同的16S rDNA序列存在共有的ST,如ST69、ST75和ST173,我们猜测这3个STs在新ST演变过程中起着重要作用。目前已有一些研究利用MALDI-TOF MS技术从蛋白指纹图谱角度对霍乱弧菌进行分析。Telesmanich等[22]证明MALDI-TOF MS可以区分霍乱弧菌O1和非O1/O139菌株,并且可以分析它们的来源和系统发育关系。由于本研究收集的菌株血清群单一(所有菌株都属于O1血清群)且地域环境相似,获得的蛋白指纹图谱聚类分析结果与核苷酸序列系统发育分析结果间未发现明显联系,下一步我们将丰富菌株血清型和地域信息,已获得更加全面的霍乱弧菌种群结构信息。
总之,本研究通过MLST对我国沿海地区192株O1群霍乱弧菌的分子分型特征分析发现,ST69和ST75是O1群霍乱弧菌最主要的2个ST,管家基因重组比突变更容易发生,O1/O139群和非O1/O139群霍乱弧菌间存在较大差异,另外利用MALDI-TOF MS技术的分型研究有待进一步探究。
利益冲突:无
引用本文格式:柳婷婷,李萍,杨文辉,等.我国主要沿海地区O1群霍乱弧菌分子分型特征分析[J].中国人兽共患病学报,2022,38(1):10-18.DOI:10.3969/j.issn.1002-2694.2021.00.177
