单增李斯特菌穿越生理屏障机制的研究进展
2022-08-09郑文亚董熙奇张亿银
高 凡,郑文亚,2,刘 犇,2,董熙奇,张亿银
单增李斯特菌(Listeriamonocytogenes, LM)属革兰阳性菌,1926年首次从被感染的兔子和豚鼠中发现,在20世纪70年代被确认为一种人兽共患疾病的病原。在80年代被确定为一种食源性病原体,是食品加工业中主要的污染源之一。该菌对恶劣环境的抵抗能力强,能够在高盐、低pH和低渗条件下生存,还能在4 ℃左右的温度下生长[1-2]。在致病方面,LM对宿主肠道屏障、胎盘屏障、血脑屏障(Blood Brain Barrier,BBB)的侵袭作用是导致感染的关键事件,感染后可使免疫力低下的动物发生细菌性败血症、脑膜炎、流产等病理症状[3]。目前虽然关于LM介导的致病性方面研究较多并取得了一定的成果,但在感染机制方面的研究较少,尤其对3种生理屏障的侵袭机制还有待深入探索。本文将对LM侵袭机制的研究进展进行综述,为进一步研究LM致病机理提供一定参考。
1 LM对不同生理屏障的侵袭
1.1 对肠道屏障的侵袭 肠道屏障是阻止肠道病原体感染的第一道防线,在抵御肠道内大量细菌及其代谢产物的入侵、维持机体内环境相对稳定方面具有重要作用。LM则能通过不同途径进入肠上皮细胞,然后依靠淋巴循环和血液循环传播至靶器官,进而介导对多个组织脏器的侵袭,如大脑、胎盘等[4]。研究表明,LM对肠道屏障侵袭方式复杂多样,可通过粘附蛋白(Listeria adhesion protein, LAP)、内化素A(Internalin A, InlA)及肠道M细胞等途径介导内化[5]。LAP、InlA均为LM表面粘附蛋白,其中LAP可通过与上皮细胞表面热休克蛋白60(Heat shock protein 60,HSP60)作用,介导胞浆中的IKK反应,激活NF-kB通路[6]。此外,NF-kB通路还将激活肌球蛋白轻链激酶(Myosin light-chain kinase, MLCK),从而导致肌球蛋白轻链(myosin light chain, MLC)磷酸化,使连接蛋白、钙粘蛋白、网格蛋白重排,进而打开紧密连接,导致LM进一步感染[7-8]。同时该过程还将导致促炎症因子TNF-α、IL6、IL8的大量分泌,诱发炎症反应。InlA由800个氨基酸构成,包含一个由15个亮氨酸构成的结构域(leucine-rich repeats,LRR)。在LRR区域的下游,存在一个重复区域,该区域对LRR区域与钙粘蛋白的结合至关重要[9]。InlA通过与钙粘蛋白(E-cadherin)直接作用介导内化,同时诱导其泛素化和磷酸化,促进钙粘蛋白的招募,提高内化效率。InlA介导的内化作用被认为是LM穿越肠道屏障的主要途径,而内化素B(Internalin B, InlB)在入侵肠道屏障过程中则不发挥作用[10]。此外,InlA与钙粘蛋白在相互作用上存在物种特异性,如InlA不能与小鼠的钙粘蛋白结合,但能够与人、兔细胞表达的钙粘蛋白结合并发生内化作用[11]。这是因为构成小鼠钙粘蛋白的第16个氨基酸是谷氨酸,而在LM感染模型中,钙粘蛋白第16位氨基酸为脯氨酸,对其N-端结构域测定结果显示脯氨酸直接参与了与InlA的相互作用。同时,LM还能在肠道杯状细胞与上皮细胞之间的交界位置侵入,从而穿越肠道屏障[12-13]。
而在以M细胞为通道的侵袭过程中,LM通过感染Peyer氏结(Peyers patch,PP)中的CX3CR1+细胞,介导并产生促炎症因子IL-23、IL-12。而IL-23将诱导固有淋巴细胞(Innate lymphocyte 3, ILC3)和γαT细胞分别产生IL-22、IL-17,其中IL-17将继续诱导gp38+基质细胞产生IL-11,而IL-11与IL-22对上皮细胞的STAT3过程具有协同促进作用[14]。此外,IL-12与ILC1细胞相互作用,将激活IFN-γ通路,从而诱导STAT1过程。而STAT3与STAT1过程促使肠上皮细胞增殖,并且提高杯状细胞中钙粘蛋白的表达量,从而使LM依赖InlA介导的内化作用增强[15-16]。另有研究表明,缺失InlA和LAP并不能显著阻止LM的内化效率,这提示在对肠道的侵袭中可能还存在其他重要的未知途径[17]。
1.2 对胎盘屏障及BBB的侵袭 胎盘屏障对于胎儿正常发育、阻止微生物入侵等方面具有重要作用。但胎盘屏障相对于肠道屏障在免疫功能方面存在较大区别,如免疫细胞的数量和种类[18]。肠道屏障存在大量免疫细胞,而胎盘屏障则缺乏,这种差异使胎盘组织对病原微生物的清除能力显著低于肠道组织,给高毒力、高侵袭的病原微生物的大量定殖和复制提供了机会[19]。
Disson O等[20]通过动物模型研究表明,LM在侵袭胎盘的过程中,InlA、InlB存在相互依赖关系。进一步研究表明,宿主细胞中PI3激酶(PI3 kinase)的活性强度是影响InlA与InlB之间协同作用的关键,以InlA的侵袭方式受到细胞内PI3激酶活性水平的影响。PI3激酶在胎盘组织细胞中活性较低,InlB可通过与肝细胞生长因子(Met)的相互作用,显著提高PI3激酶的活性水平,从而导致肌动蛋白骨架重排,使InlA得以完成对胎盘细胞的入侵[21]。同时,InlA无法独立完成对胎盘细胞的侵袭,而InlA与钙粘蛋白的相互作用是InlB发挥作用的先决条件[22]。另有研究表明,LM分泌蛋白InlP在介导胎盘侵袭过程中同样具有重要作用,InlP是LM产生的一种分泌蛋白,能够与胎盘组织中的丝状肌动蛋白(Afadin)结合,使LM介导对胎盘组织的侵袭[23]。与此同时,InlP与丝状肌动蛋白之间的作用强度与Ca2+浓度相关,胞质中的Ca2+浓度变化可能导致InlP构型发生变化,通过影响InlP的活性增强InlP与丝状肌动蛋白结合能力,这可能与InlP结构中132-136氨基酸之间的环形结构与Ca2+存在一定的亲和力有关[24]。丝状肌动蛋白定位于胞间连接处,在细胞表面并不表达,而在含有核蛋白的细胞间连接的胞质中表达丰富,同时在维持细胞极化、迁移等方面发挥重要作用[25]。研究表明,胎盘组织细胞中PI3激酶活性水平显著低于肠道细胞,而在PI3激酶高表达的肠道细胞中InlA就足以介导内化,这解释了内化素B为何在对肠道的侵袭中不发挥作用[26-27]。LM在肠道和胎盘屏障中的内化差异,显示胎盘组织对抵御病原菌侵袭具有天然、高效的保护作用,反映了哺乳动物在应对病原菌感染存在协同进化特点。见图1。
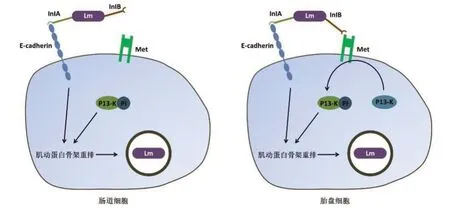
图1 InlA与InlB在肠道组织和胎盘组织细胞中的协同作用差异
BBB维持着脑部微环境的动态稳定,紧密连接、脑毛细血管网和极化的微血管内皮细胞共同维持了BBB结构和功能的完整性,同时也是抵御病原微生物侵入的关键屏障[28]。LM可通过血液循环和淋巴循环介导对BBB的定殖和侵袭,从而导致严重的中枢神经系统疾病。研究表明,InlA与InlB在对BBB的侵袭过程中也可能存在相互依赖关系,但这种依赖作用是否受到PI3激酶活性的影响还需要进一步研究[29]。此外,在BBB多种细胞上发现HSP60的表达,这提示LAP也可能在侵袭过程中发挥作用。另有研究表明,内化素F(Internalin F,InlF)通过与大脑内皮细胞表面的波形蛋白(Vimentin)相互作用,从而对内皮细胞进行粘附和内化[30]。见表1。
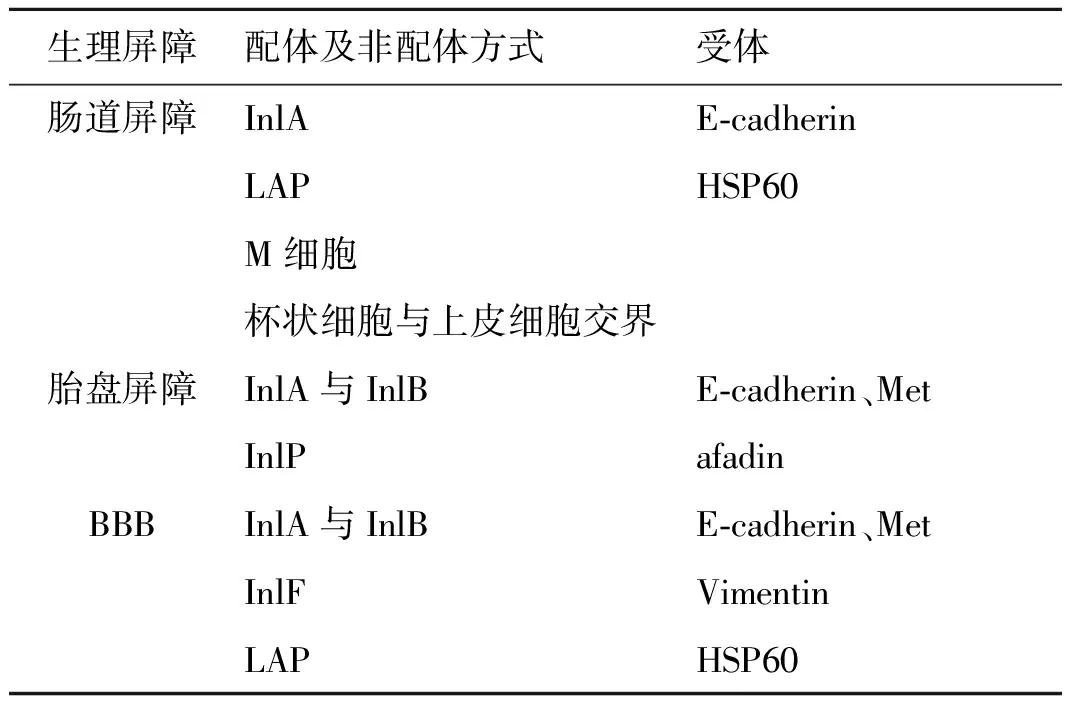
表1 单增李斯特菌在不同生理屏障中内化素与受体的种类及作用方式
2 LM在细胞内及细胞间的传播
LM在感染非吞噬细胞后,将通过分泌多种毒力因子如溶血素O(LLO)、磷脂酶A(PlcA)、磷脂酶B(PlcB)、LntA介导胞内逃逸和胞间传播,从而导致细胞损伤[31]。研究表明,LLO可介导内质网应激、改变溶酶体的通透性、影响线粒体的正常形态,诱导内质网中Ca2+的释放及细胞膜的成孔反应,使细胞内外Ca2+和K+浓度失调,胞内Ca2+浓度升高,胞外K+浓度升高,进而影响细胞的正常生理功能,而PlcA与PlcB 则会与LLO进行协同作用[32-33]。Li TL等[34]研究发现,LLO可进一步诱导线粒体上的mtCa2+转运体(mtCa2+uniporter,MCU)通道过度开放,加速对Ca2+的摄取。同时线粒体中的高钙环境将提高丙酮酸脱氢酶(PDH)的活性,而PDH是乙酰辅酶A(CoA)生成的关键酶,从而提高了CoA的生成量。过量形成的CoA将使Rubicon蛋白乙酰化,Rubicon是LC3相关吞噬作用(LC3-associated phagocytosis,LAP)的重要蛋白,LAP是宿主细胞吞噬和降解病原体的过程[35]。所以,被乙酰化的Rubicon蛋白将显著影响LAP过程,从而使LM躲避杀灭,实现胞内生存和扩散[36]。此外,Zhang YF等[37]研究发现在LM胞内逃逸的过程中,线粒体通过产生超氧化物、过氧化氢以及释放含有活性氧(ROS)的囊泡,抑制其繁殖并杀灭细菌。但抑制线粒体自噬,ROS的生成显著增加,反之则明显减少。另有研究表明,LLO通过介导线粒体自噬受体NLRX1寡聚化,使NLRX1中含有的LC3相互作用区域(LC3-Interacting region,LIR)与LC3结合,诱导线粒体自噬,从而消除线粒体产生的杀菌物质对其生存的影响[38]。见图2。
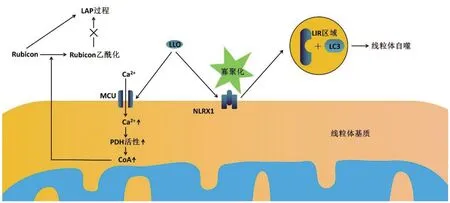
图2 LLO与线粒体的相互作用
此外,LntA通过与核内蛋白BAHD1(Bromo adjacent homology domain-containing 1 protein , BAHD1)的相互作用,使调控炎症因子与干扰素分泌的ISGs基因被抑制,从而影响细胞对LM的免疫反应,介导免疫逃逸[39]。LM在胞质运动的过程中,肌动组装蛋白(ActA)与肌动蛋白核化复合体2/3(Actin nucleated complex 2/3, ARP2/3)共同作用,ARP2/3复合物由7个亚基组成:ARP2、ARP3和ARPC1-5,它们共同介导肌动蛋白的成核和分支,与ActA一同促进肌动蛋白的聚合[40]。肌动蛋白聚合并产生足够的动力在胞质中穿梭,最后在InlC的协助下实现细胞间的传播[41]。InlC是LM产生的一种分泌蛋白,属于细菌内化素蛋白家族成员之一,其表达受到毒力调节蛋白(Virulence regulatory protein)的调控,在内化后的宿主细胞内高度表达。同时,InlC在宿主细胞中具有多种功能,包括对细胞免疫反应的刺激、诱发炎症因子的产生[42]。InlC通过改变极化上皮细胞的顶端连接形态来调节突起的形成,在正常生理过程中,胞间极化上皮细胞顶端张力的形成需要依靠哺乳动物适配器蛋白(Tuba)和肌动蛋白调控蛋白(N-WASP2)的结合,该结合过程发生在Tuba羧基端SH36结构域,形成的张力效应会抑制细菌的穿胞过程[43-44]。而InlC则可能通过削弱N-WASP2的活性,或通过与N-WASP2竞争性结合Tuba,降低N-WASP2与Tuba的结合效率,从而增强LM在胞间形成突起的能力[45]。同时InlC与Tuba的结合将会使原本绷紧的顶端连接变的舒展,易形成突起,利于胞间传播。而在感染缺乏极化能力的细胞系时,可能仅依靠ActA聚合肌动蛋白产生的推动力介导质膜突起的形成,该过程中是否依赖InlC的介导存在争议[46]。LM感染的大部分组织细胞具有极化能力,极化细胞能使组织形成致密的屏障并产生上皮张力,而张力作用可能诱导InlC响应机制被触发。见图3。
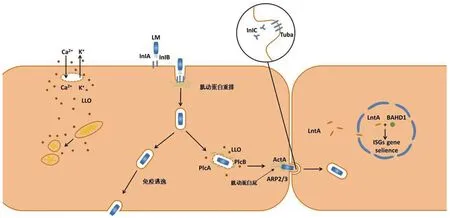
图3 LM在细胞内逃逸及细胞间传播的主要作用机制
3 展 望
虽然近年来在探究LM对不同生理屏障的侵袭机制方面已取得了很大进展,但仍有一些重要问题有待探索。例如:LLO引起的胞质Ca2+浓度变化如何影响InlP的功能?LLO的成孔效应使胞内外Ca2+浓度失衡,胞内高Ca2+如何影响InlP功能?这些都需要进一步确定。而通过鉴定Ca2+与InlP作用形成复合物的分子结构有望揭示其作用机制。InlF与大脑内皮细胞表面波形蛋白作用引起的胞内反应也有待阐明,这将有助于理解LM对BBB和胎盘屏障侵袭的细胞作用特点。
在胞内过程中,LntA通过与BAHD1相互作用来抑制ISGs基因的功能,进而降低机体对LM的免疫水平,协助免疫逃逸。尽管ISGs基因的部分功能已被发现,但是LntA诱导BAHD1对ISGs抑制作用的分子机制尚不清晰。同时上皮细胞张力与InlC之间的响应机制也有待研究,包括InlC在穿胞过程中是否参与了细胞骨架的调控。以上问题的解决对于全面理解LM的侵袭机制及胞内作用具有重要意义。
总之,LM通过细胞受体介导入胞及胞内作用是其致病性的关键原因,未来在该方向上利用转录组学与蛋白质组学研究方法的相互配合,有望进一步理解和补充LM致病性的内在机制。
利益冲突:无
引用本文格式:高凡,郑文亚,刘犇,等.单增李斯特菌穿越生理屏障机制的研究进展[J].中国人兽共患病学报,2022,38(1):55-61.DOI:10.3969/j.issn.1002-2694.2021.00.173
