超高效液相色谱-串联质谱法快速检测麦类中典型链格孢霉毒素
2022-07-07邢家溧郑睿行张爱芝毛玲燕徐晓蓉娄永江穆应花
吴 希,邢家溧,郑睿行,张爱芝,毛玲燕,徐晓蓉,娄永江,穆应花
(1.宁波大学食品与药学学院,浙江 宁波 315211;2.宁波市产品食品质量检验研究院(宁波市纤维检验所),浙江 宁波 315048)
链格孢霉毒素(toxins,ATs)是链格孢霉菌产生的次级代谢物,最为典型的有链格孢酚(alternariol,AOH)、交链格孢酚单甲醚(alternariol monomethyl ether,AME)、交链孢烯(altenuene,ALT)、细交链格孢菌酮酸(tenuazonic acid,TeA)、交链孢毒素I(altertoxin I,ATX-I)、腾毒素(tentoxin,Ten)和细格菌素(altenusin,ALS)。这些毒素在自然界中广泛存在,会侵染谷物导致作物减产,还能污染后续的加工产品造成经济损失。ATs具有急慢性毒性以及三致效应(致癌、致畸、致突变),对人体健康构成严重威胁。
麦类作物易受ATs污染,Siegel等在荞麦粉中检出TeA含量高达851 μg/kg;何玲等在调查四川省市售小麦及其制品中ATs污染情况时,在小麦及其制品检出AOH、AME、TeA和Ten含量分别为0.75、0.57、27.2 μg/kg和4.78 μg/kg;Asam等在调查婴儿谷物产品中的ATs污染情况时,在小麦、燕麦、黑麦产品中检出TeA含量为8~30 μg/kg。随着ATs危害逐渐明晰,人们的风险防范意识也逐步加强,但因缺乏准确的检测分析方法和系统性的风险评估数据,鲜见ATs的相关法规与限量标准,目前我国仅有1 项ATs的行业标准,但应用范围只限于部分果蔬。因此,要尽快建立简单、快速、高效、灵敏的ATs检测方法,推动我国麦类中ATs残留水平的控制、检测标准的制定和管理措施的采取。
目前,ATs的主要检测技术有薄层色谱(thin layer chromatography,TLC)法、酶联免疫检测(enzymelinked immunosorbent assay,ELISA)法、气相色谱(gas chromatography,GC)和气相色谱-串联质谱(gas chromatography-tandem mass spectrometry,GC-MS/MS)法、高效液相色谱(high performance liquid chromatography,HPLC)法和超高效液相色谱-串联质谱(ultra-high performance liquid chromatographytandem mass spectrometry,UPLC-MS/MS)法等。TLC与ELISA法操作繁琐且灵敏度低,ATs稳定性好而挥发性差在GC和GC-MS/MS检测方法中受到限制。UPLC-MS/MS具有高灵敏度及高选择性等优点,近年来对ATs的分析主要集中在UPLC-MS/MS的研究和应用方面。但目前麦类中ATs的研究大多集中于单一或几种毒素的检测,如Siegel等利用UPLC-MS/MS检测荞麦粉中TeA,检出限为10 μg/kg;何玲等利用HPLC结合荧光检测器同时检测小麦中的AOH、AME、TeA、Ten毒素,检出限为0.2~1.0 μg/kg;Asam等用UPLC-MS/MS法测定小麦、燕麦、黑麦中的TeA,检出限为1 μg/kg。另外,为了进一步提高检测效果,目前报道了多种ATs的提取和富集方法,包括QuEChERS法和固相萃取方法。但这些方法虽然前处理简单,灵敏度高,但检出限高,只能同时检测少数几种ATs。
基于以上研究现状,本研究拟采用具有操作简单、富集倍数高、有机溶剂用量少、成本低廉等优点的分散液-液微萃取(dispersive liquid-liquid microextraction,DLLME)方法,快速富集和净化麦类中7种ATs,再结合UPLC-MS/MS技术,实现麦类中7种ATs的快速定性和定量测定。这将为麦类中ATs的污染状况研究提供方法依据,也为我国食品中ATs限量标准的制定提供技术支持。
1 材料与方法
1.1 材料与试剂
麦类样品购于当地超市,磨碎(500 r/min,5 min),过筛(40 目),于0 ℃条件下保存。
甲醇、乙腈、甲酸、乙酸乙酯、氯苯、二氯甲烷、三氯甲烷(均为色谱纯) 美国Sigma公司;无水硫酸镁、氯化钠(均为分析纯) 国药集团化学试剂有限公司;毒素标准品:AOH、AME、ALT、TeA、ATX-I、Ten和ALS(纯度均≥98%) 上海安谱实验科技股份有限公司。
1.2 仪器与设备
Xevo TQ-XS三重四极杆质谱仪、BEH C色谱柱(50 mm×2.1 mm,1.7 μm)、HSS T色谱柱(50 mm×2.1 mm,1.8 μm) 美国Waters公司;X1R高速离心机 美国赛默飞世尔科技公司;TGL-20M高速台式冷冻离心机 上海卢湘仪离心机仪器有限公司;TurboVap® LV多功能全自动样品浓缩仪 上海Biotage有限公司;Milli-Q型超纯水机(电阻率为18.2 MΩ·cm)美国Millipore公司;IKA-Vortexd2旋涡混合器 德国IKA(艾卡)仪器设备有限公司;Multi Reax-Heidolph多管漩涡振荡器 德国海道夫仪器设备有限公司;ME-204电子分析天平(精度为0.000 1 g) 梅特勒-托利多仪器有限公司;KS-300EI超声波清洗机 宁波科生设备有限公司。
1.3 方法
1.3.1 溶液配制
标准储备液:分别准确称取AOH、AME、ALT、TeA、ATX-I、Ten和ALS标准品0.001 g(精确至0.000 1 g)溶于10 mL乙腈中,配制成质量浓度为100 mg/L的标准混合液,取100 μL标准混合液(100 mg/L),并用乙腈溶解定容至10 mL棕色进样瓶中,得到质量浓度为1 mg/L的标准储备液,密封后置于-20 ℃保存、备用。
标准工作液:用乙腈-水(1∶1,/)将标准储备液逐级配制成质量浓度分别为0.5、1、2、5、10、20、50、100 μg/L的7种ATs的混合标准溶液。
基质标准工作液:用空白基质溶液逐渐稀释1 mg/L的混合标准储备液,制备成10 μg/L的基质标准工作液。
1.3.2 提取和净化
准确称取1 g样品(精确至0.01 g),置于50 mL的螺口尖底离心管中,加入3 mL一级水、10 mL1.5%甲酸乙腈-甲醇(4∶1,/)提取剂,涡旋3 min。加入2 g无水MgSO、1 g NaCl在40 ℃下超声5 min、振荡提取15 min,9 500 r/min、4 ℃离心10 min,取1 mL上清液。
加入100 μL三氯甲烷萃取剂、5 mL的1.5 g/L氯化钠溶液、0.4%甲酸溶液(/),4 500 r/min离心5 min,取下清液,氮吹浓缩至干,用甲醇溶液复溶至1 mL,过0.22 μm有机滤膜,待UPLC-MS/MS分析。
1.3.3 分析条件
BEH C色谱柱(50 mm×2.1 mm,1.7 μm);柱温为40 ℃;流动相:A为0.1%甲酸溶液(/),B为乙腈;流速0.40 mL/min;进样体积5 μL;梯度洗脱程序:0~5.0 min,90%~5% A、10%~95% B;5.0~7.0 min,5% A、95% B;7.0~7.5 min,5%~90% A、95%~10% B;7.5~10.0 min,90% A、10% B。
电喷雾离子源(electrospray ionization,ESI),正负离子模式扫描;毛细管电压1.08 kV;锥孔电压25 V;射频透镜1和射频透镜2的电压均为15.0 V;离子源温度150 ℃;脱溶剂温度600 ℃;脱溶剂气流量1 000 L/h;锥孔反吹气流量150 L/h;多反应监测(multiple reaction monitoring,MRM)模式。7种ATs的保留时间和质谱参数见表1。

表1 MRM模式下7种ATs的保留时间和质谱测定参数Table 1 Retention time and mass spectral parameters for the seven ATs in MRM mode
2 结果与分析
2.1 仪器条件的优化
2.1.1 质谱条件的优化
取7种目标物的混合标准溶液(100 μg/L),分别以自动进样的方式在全扫检测模式下进行质谱条件优化。结果显示ATX-I在ESI模式扫描下不成峰,而在ESI模式扫描时响应值较高(图1),且峰形尖锐,剩下6种ATs在ESI模式得到的离子峰的响应值更佳。通过子离子扫描得到目标物碎片化离子信息,选择最强的产物离子和次强的产物离子分别作为7种ATs的定性和定量离子,并对锥孔电压、碰撞电压、离子源温度、脱溶剂气体温度、碰撞气体流量质谱参数进行优化。实验优化的质谱条件,使每种靶物质得到最佳电离效率(表1)。
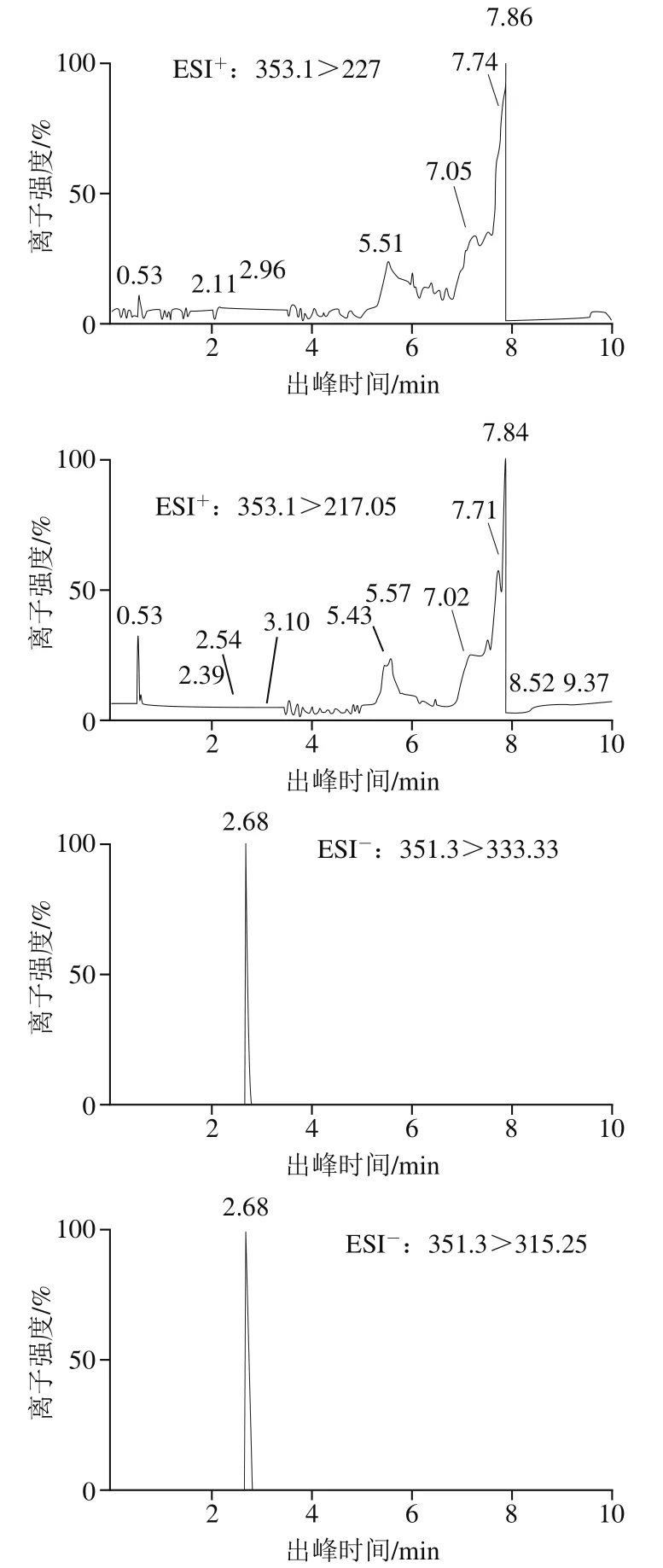
图1ATX-I毒素的离子对质谱图Fig. 1 Ion pair mass spectra of ATX-I
2.1.2 色谱条件的优化
分别选择纯水-乙腈和0.1%甲酸-乙腈溶液(/)作为流动相,比较2个流动相体系对分离效果的影响。结果发现,在纯水流动相中TeA毒素会出现拖尾现象,而纯水加入0.1%甲酸溶液(/)有利于抑制真菌毒素与色谱柱的静电作用,避免拖尾现象,TeA毒素峰形对称性明显得到改善。因此本实验最终选择0.1%甲酸-乙腈溶液作为流动相。
取质量浓度为100 μg/L的7种目标物混合标准溶液,以0.1%甲酸-乙腈溶液作为流动相,按表1进行梯度洗脱,在保持浓度、流速、进样量等参数一致的条件下,考察HSS T(50 mm×2.1 mm,1.8 μm)和BEH C(50 mm×2.1 mm,1.7 μm)这2种色谱柱对7种ATs的分离效果以及每种毒素的峰形、保留时间。结果显示BEH C分离效果优于HSS T,7种ATs在BEH C柱上能获得更好的峰形和检测灵敏度,且BEH C具有广谱性、次级相互作用较低,pH值耐受范围宽(pH 1~12),柱效更高,峰形尖锐等优点,故选择BEH C作为分析柱。
同时对流速、柱温及梯度洗脱程序进行优化,确定流速为0.4 mL/min;柱温为40 ℃;由于7种ATs具有不同的极性,ALT的极性较强,AME的极性较弱,为了兼顾7种ATs能同时出现良好峰形,需要对流动相进行较大的梯度变化,通过不断调整,确定的洗脱程序见1.3.3节。在最优条件下得到的7种ATs色谱图见图2,可以看出7种ATs在5 min内可较好分离。

图2 优化条件下6种ATs的正离子图(A)和ATX-I的负离子图(B)Fig. 2 Positive ion chromatograms of six ATs (A) and negative ion chromatogram of ATX-I under the optimized conditions (B)
2.2 提取液的优化
ATs易溶于甲醇与乙腈,其中TeA的酸性和极性较强,需在提取剂中加入适量的酸,1.5%甲酸最有利于TeA的提取。研究比较1.5%甲酸-乙腈(/)、1.5%甲酸乙腈-甲醇的不同比例对7种ATs提取效果的影响,结果表明,添加甲醇对ATX-I的提取效果影响明显,对比1.5%甲酸-乙腈(/)提取体系,ATX-I的回收率在1.5%甲酸乙腈-甲醇(4∶1,/)体系中提升10%,这也与畅彤等研究适量的甲醇有利于ATX-I提取一致;甲酸的添加有利于TeA的提取,在1.5%甲酸-乙腈(/)提取体系中回收率高达92%;1.5%甲酸乙腈-甲醇比例从1∶4到4∶1,小麦样品的提取液颜色由深黄色变为浅黄色,猜测色素这一干扰物对实验的干扰在减小。1.5%甲酸乙腈-甲醇(4∶1,/)提取体系中7种ATs的回收率最佳,在78.7%~100%之间(图3),故选择1.5%甲酸乙腈-甲醇(4∶1,/)提取体系。
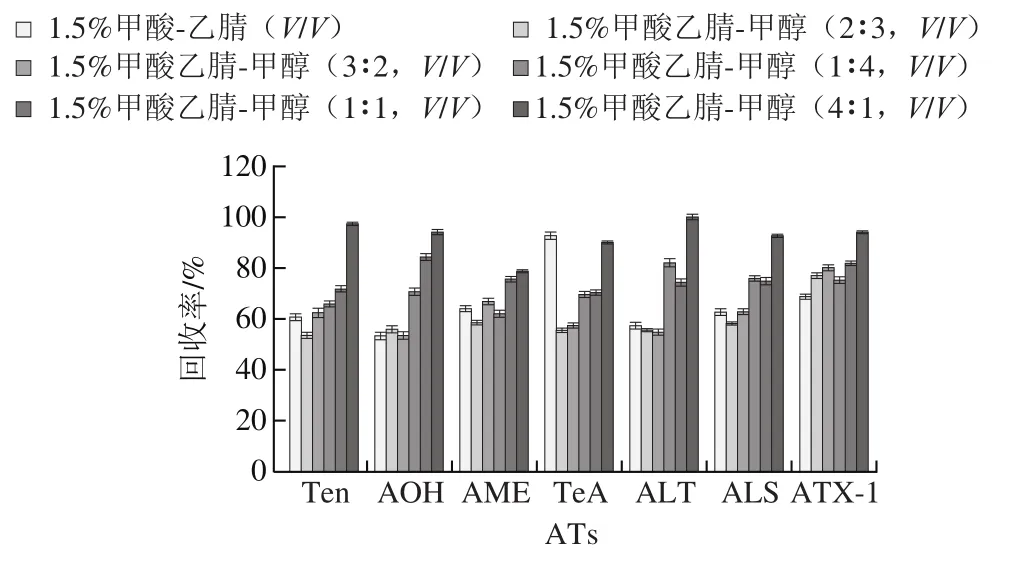
图3 提取溶剂体系对小麦样品中7种ATs回收率的影响(n=5)Fig. 3 Effect of extraction solvent systems on the recoveries of seven ATs in wheat samples (n = 5)
2.3 萃取条件的优化
2.3.1 萃取剂的优化
小麦样品经提取、盐析分层后,蛋白质、油脂、色素以及糖组分等会被萃取出来,为减小杂质的影响,对萃取条件进行考察。以1.5%甲酸乙腈-甲醇(4∶1,/)提取液作为分散剂,分别考察乙酸乙酯、氯苯、二氯甲烷、三氯甲烷作为萃取剂时对各目标组分的萃取效率,以7种ATs含量计算回收率。使用乙酸乙酯作为萃取试剂时没有观察到沉积相,因此不适合作为萃取试剂。使用其他3种萃取剂所得回收率见图4,三氯甲烷回收率(83.1%~94%)比氯苯(75.1%~88.7%)和二氯甲烷(62.7%~72.7%)较优,当以三氯甲烷作为萃取剂时,各个目标物质的萃取效率较好,对目标组分无干扰,且三氯甲烷在水中的溶解度较小,可以减少萃取剂的损失,故选择三氯甲烷作为本实验的萃取溶剂。
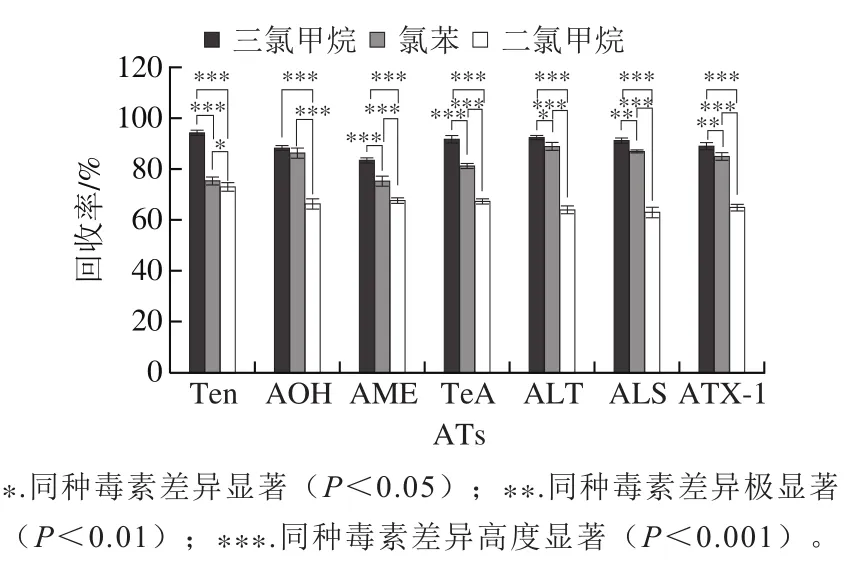
图4 萃取剂种类对7种ATs回收率的影响(n=5)Fig. 4 Effect of extractant type on the recoveries of seven ATs in wheat samples (n = 5)
2.3.2 萃取体积的优化
由于使用三氯甲烷溶剂进行DLLME时,小麦样品基质中某些成分会沉淀在离心管底部,影响萃取试剂的吸取。因此,使用更大体积的萃取试剂可以更方便吸取离心后的萃取试剂。在分散剂体积为1 mL的条件下,分别用50、100、150 μL的三氯甲烷进行萃取,考察不同体积萃取剂对萃取回收率的影响。结果如图5所示,随着三氯甲烷体积增大,回收率也增大,而当体积大于100 μL时,回收率增加不明显,而且三氯甲烷的使用体积越大,实验更耗时,最终萃取剂中三氯甲烷的占比也越大,对于含有甲酸的流动相体系,容易造成溶剂效应,使峰形变差。因此,最终确定三氯甲烷100 μL为最佳萃取体积。

图5 萃取剂体积对7种ATs回收率的影响(n=5)Fig. 5 Effect of extractant volume on the recoveries of seven ATs in wheat samples (n = 5)
2.4 方法学验证
2.4.1 方法的线性范围、检出限、定量限
以乙腈为溶剂,配制7种ATs系列混合标准溶液直接进样分析。以质量浓度为横坐标(,μg/L),以峰面积为纵坐标(,AU),绘制目标分析物的基质加标校正曲线(表2)。结果显示,7种ATs在各自范围内均能获得良好的线性关系,均大于0.987 277,以信噪比3和信噪比10确定方法的检出限和定量限。7种ATs的检出限和定量限分别为0.11~0.16 μg/kg和0.42~0.49 μg/kg。

表2 7种ATs的线性范围、相关系数、检出限和定量限Table 2 Linear ranges, correlation coefficients, detection limits, and quantification limits of seven ATs
2.4.2 加标回收率以及精密度
按1.3.2节处理小麦样品,分别以1、2 倍和10 倍定量限进行三水平加标实验。每个水平在1 d内重复测定5 次,计算平均回收率和相对标准偏差(relative standard deviation,RSD)。如表3所示,7种ATs的平均加标回收率为70.7%~101.3%,RSD为1.22%~4.11%,均小于10%,说明该方法性能良好。本方法对小麦样品的不同含量的7种ATs均具有较高的回收率和精密度,满足检测要求。
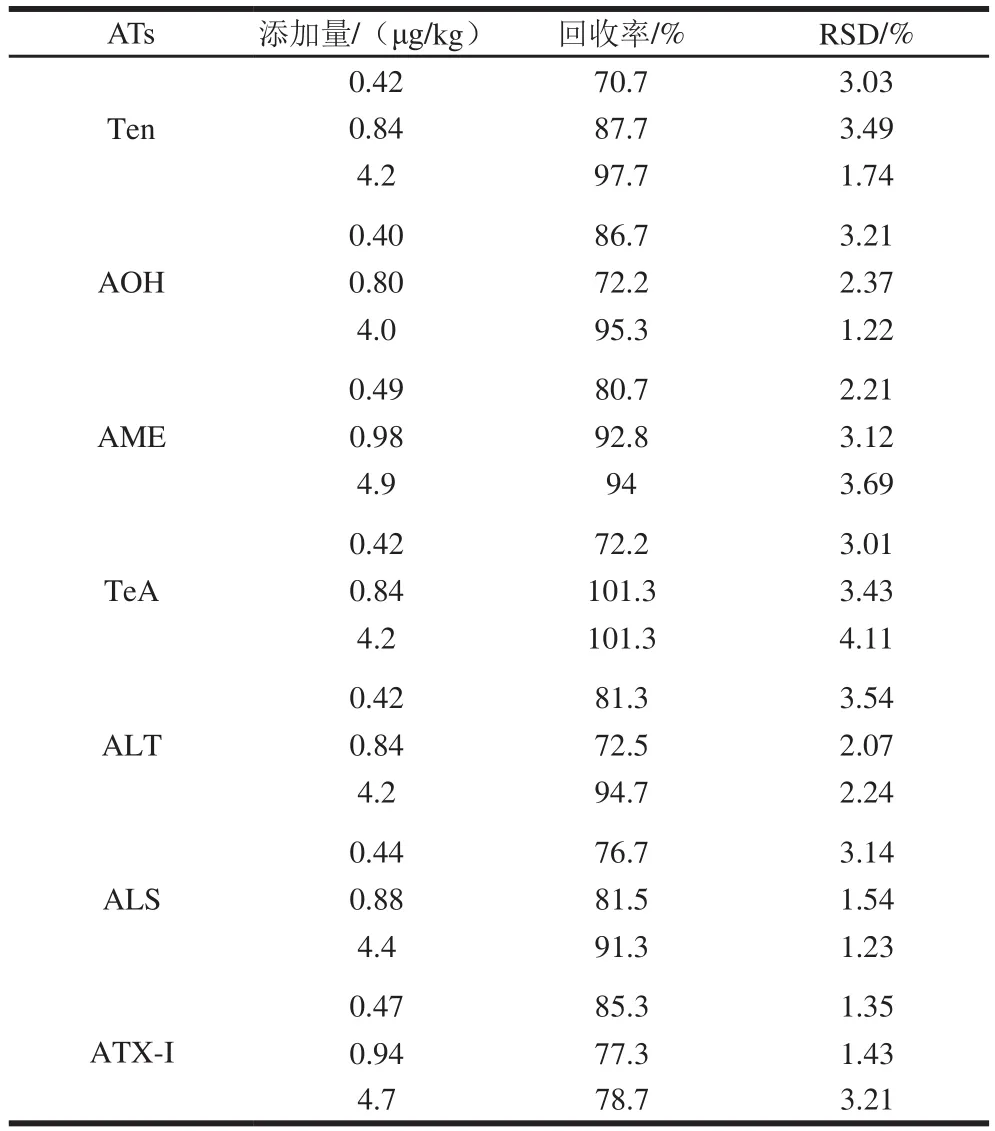
表3 7种ATs在小麦样品中的加标回收率和精密度(n= 5)Table 3 Recoveries and precision of seven ATs spiked in wheat samples (n = 5)
2.4.3 基质效应
取质量浓度为10 μg/L的7种目标物混合标准溶液,比较基质提取液和纯溶剂中7种ATs响应值,以评估基质效应(matrix effect,ME)。ME<85%或ME>115%,存在基质抑制或增强作用,85%≤ME≤115%,基质对分析物的分析过程无显著的干扰。
如表4所示,小麦样品中7种ATs的ME在90.7%~115%之间,基质对分析物的分析过程无显著干扰,保证了检测结果的准确度与可靠性。

表4 7种ATs在小麦样品基质的ME(n=5)Table 4 Matrix effect of seven ATs in wheat samples (n = 5)
2.4.4 与其他方法比较
如表5所示,本研究与QuEChERS方法相比回收率高,比固相萃取处理步骤简单、试剂用量少、成本低廉等,且本研究涉及的ATs种类广、数量多,结合UPLC-MS/MS具有检测灵敏度高、抗干扰能力强、定性准确等优点,能用于麦类样品中7种ATs的快速(5 min内)定性、定量分析测定。

表5 本实验方法与其他文献方法的比较Table 5 Comparison of the UPLC-MS/MS method and other existing methods present in the literature
2.4.5 实际样品测定
使用本实验建立的方法对市购黑麦、荞麦、莜麦、青稞、燕麦、小麦6种麦类样品(各20个)进行7种ATs的检测分析,结果见表6。结果显示这6种麦类中7种ATs均有不同程度的检出,黑麦样品检出Ten、AOH、ALT、TeA、ALS;荞麦样品中检出Ten、ALT、TeA、ALS、ATX-I;莜麦样品中检出7种ATs;青稞样品中检出Ten、ALT、TeA、ALS、ATX-I;燕麦样品中检出Ten、AOH、TeA、ALS、ATX-I;小麦样品中检出Ten、AOH、AME、ALT、TeA、ALS。Ten和TeA是6种麦类中都检出的毒素,含量分别是0.6~10.7 μg/kg和未检出~31 μg/kg;ALS虽检出率低于Ten和TeA,但最高含量37.3 μg/kg与TeA相当。实际样品测定结果表明Ten、TeA广泛存在于麦类中,这一研究结果与之前研究报道的结果基本一致。

表6 麦类样品中7种ATs的含量统计Table 6 Statistical analysis of results obtained for the determination of seven ATs in rye, buckwheat, naked oat, barley, oat, and wheat

续表6
3 结 论
采用DLLME前处理结合UPLC-MS/MS同时检测麦类中7种ATs的分析方法。该方法具有操作简单、快速(7种ATs在5 min内完成色谱分离分析)、回收率高、灵敏度高、检出限低等优点,能满足麦类中真菌毒素的检测要求,具有实际应用价值。将该方法应用于麦类样品中的实际测定,结果显示Ten和TeA是6种麦类中都检出的毒素,含量分别为0.6~10.7 μg/kg和未检出~31 μg/kg;ALS虽检出率低于Ten和TeA,但最高含量37.3 μg/kg与TeA相当,这些结果将为粮食中ATs残留水平的控制、检测标准的制定和管理措施的采取,都具有重要的理论和现实意义。
