食用级淀粉纳米颗粒乙酰化疏水改性及其消化特性
2022-07-07姚先超史永桂焦思宇吴雅珊杨婉颜林日辉
姚先超,史永桂,焦思宇,吴雅珊,杨婉颜,林日辉
(广西民族大学化学化工学院,林产化学与工程国家民委重点实验室,广西林产化学与工程重点实验室/协同创新中心,广西多糖材料与改性重点实验室,广西 南宁 530006)
近年来,纳米颗粒材料作为药物缓释载体被认为是提高药物稳定性、生物利用率的重要途径。生物质纳米颗粒也被认为是用于替代传统乳化剂、抑制油滴聚集凝结、用制备更安全环保更稳定的药用食用性Pickering乳液、饮品等的重要乳化稳定剂原料。来源广泛的纳米淀粉因其天然可再生、无毒性、生物相容性好、可生物降解性、成本低等优势,成为制备新型纳米颗粒的生物质资源。然而天然的纳米淀粉由于其组成结构中含有大量的羟基基团而具有极强的亲水性,当暴露于水中或潮湿环境时易受水分侵蚀而很快失去完整性和耐久性,大大降低其机械性能和尺寸稳定性。此外,由于这些羟基的作用,淀粉分子具有很强的分子间和分子内氢键,容易在有机介质中发生自聚集、团聚、聚沉,因而不容易分散在油相中或油水界面,不具备形成良好稳定Pickering乳液的能力,不适用于疏水性需求的领域,需要使用物理或化学方法改变其亲疏水性能。淀粉的化学疏水改性主要有醚化、酯化、交联、接枝和缩合反应等。酯化改性是将葡萄糖单元上的活性羟基转化为烷基或芳基,这种改性不会破坏其链的完整性,保持了淀粉原有生物可降解等性能,但疏水性、加工性和柔韧性得到了改善。乙酰酯化是最容易实现淀粉疏水改性的酯化修饰方法,乙酰淀粉也是食品应用中使用非常广泛的改性淀粉。
乙酰化纳米淀粉的制备方法中,原淀粉与醋酸酐要在碱性或强酸催化下制备成酯化淀粉,再将产物溶解于有机溶剂(如二甲基亚砜、丙酮等),最后通过高速均质技术制备纳米淀粉颗粒。Teodoro等报道了用冰醋酸和乙酸酐在40 ℃下将淀粉溶解成均相状态,然后用浓硫酸作为催化剂在60 ℃下乙酰酯化,制得的乙酰淀粉用丙酮溶解后在水中沉析获得500 nm左右的淀粉纳米颗粒(starch nanoparticles,StNP)。Leonardo、Getahun等用NaOH作为催化剂合成乙酰淀粉,然后用丙酮、氯仿、二氯甲烷溶解再纳米化。在这种方法中,酯化和纳米化过程需要使用强酸碱性试剂、非环保试剂,需要反复糊化、溶解、破碎、干燥等高耗能操作。另一种常用方法是,通过酸解等方式制备成淀粉纳米晶或者StNP,再催化乙酰化得到产物。Takkalkar等用淀粉纳米晶在浓硫酸催化下在50 ℃与乙酸酐反应5 h获得取代度(degree of substitution,DS)为0.99的乙酰纳米晶产物。Qian Xiaoli等用类似的方法改性淀粉纳米晶,产物能够用于稳定Pickering乳液。Xiao Huaxi等用浓硫酸催化乙酰酯化制备了DS为2.72的产物,用于膜功能的慢速蛋白质递送。对于纳米化再改性,相比于原生淀粉颗粒,StNP和纳米晶粒粒径降低至1 μm甚至数百纳米以下,使更多的羟基活性基团在没有溶胀或者糊化的情况下暴露在颗粒表层,其更短的孔道长度使得内部的羟基也有更多的机会与试剂分子接触发生反应,使改性更容易进行,而纳米淀粉粒的葡萄糖骨架结构没有发生根本改变,仍然具有生物相容性、可生物降解等特性。但是由于颗粒内的淀粉链之间氢键的作用使其结构紧密,不易溶于冷水,试剂难以接近淀粉骨架上大部分的基团,使改性工艺较为复杂,甚至需要使用腐蚀性、毒性较强的溶剂和催化剂,这些化学试剂在食品中存在使用限制性。为此,本实验探索一种除了添加乙酸酐作为酰化试剂,不添加强酸及有害试剂的StNP表面酯化疏水改性的简便工艺条件,旨在为疏水性StNP制备寻找新型技术路线。
1 材料与方法
1.1 材料与试剂
食品级木薯淀粉由广西岑溪市三角淀粉责任有限公司无偿捐赠。
乙酸酐、盐酸(均为分析纯) 廉江市爱廉化试剂有限公司;无水乙醇、氢氧化钠(均为分析纯) 成都市科隆化学品有限公司;氘代二甲基亚砜(同位素标准品) 美国Cambridge Isotope Laboratories公司;大豆油(食用油) 益嘉海里公司;其他所有分离用溶剂均为国产分析纯。
1.2 仪器与设备
JY92-IIN超声波细胞粉碎机 宁波新芝生物科技股份有限公司;SDS350整体倾斜接触角测量仪 东莞市晟鼎精密仪器有限公司;SUPRA 55 Sapphire场发射扫描电子显微镜 德国Carl Zeiss公司;Avance600MHz型核磁共振 德国Bruker公司;MAGNA-IR550红外光谱仪美国Thermo Fisher公司;Zetasizer Nano ZS激光纳米粒度、Zeta电位分析仪 英国Malvern公司;UItime IV型X射线衍射仪 日本Rigaku公司;MiniFlex600型X射线衍射仪 日本理学公司。
1.3 方法
1.3.1 StNP乙酸酯的制备
采用本课题研发的纳米化技术通过超声破碎处理和乙醇沉降的方法制备StNP,再进行颗粒表面改性,得到产物。将食用木薯淀粉放入水中在90 ℃下完全糊化得到质量分数为3.5%的淀粉糊,1 000 W超声对淀粉溶液处理20 min,而后在剧烈搅拌下将淀粉溶液滴加到无水乙醇(1∶9,/)中,醇沉析出StNP,冻干可以获得干燥颗粒。简单的表面处理后得到的StNPp均匀分散在乙酸酐,在常温下搅拌1 h,将温度缓慢提高到75 ℃,补加乙酸酐(淀粉中葡萄糖单元与乙酸酐物质的量比为1∶3,下称料液比),继续在冷凝回流下反应2 h。反应结束后,冷却至室温,在8 000 r/min转速下离心,用无水乙醇洗涤数次,沉积物加入适量水(1∶1,/),转至宽口玻璃皿,去冻干,获得StNP乙酸酯(acetate of starch nanoparticles pretreated,StNPpAc)。
按照上述合成步骤和条件,使用同等量的原淀粉和未经表面预处理的StNP进行乙酰化,所得产物标记为StAc和StNPAc。每一种淀粉样品在相同条件下分别重复做平行样品。
1.3.2 酯化淀粉DS测定
参考文献[19],并根据实际情况作改进。取适量酯化淀粉,放入150 mL的锥形瓶中并加入50 mL的0.5 mol/L氢氧化钠溶液,在磁力搅拌器上60 ℃糊化皂化1 h。将糊化后的溶液冷却至室温,滴加2~3 滴酚酞作为指示剂,再用0.5 mol/L的盐酸进行滴定至终点,记录消耗HCl的体积数。滴定后的淀粉糊化浑浊液,在60 ℃减压蒸发水分,残渣在80 ℃真空干燥至恒质量,记录残渣质量。按式(1)、(2)计算DS:

式(1)中:为乙酰基在淀粉酯中的含量/(g/g);为滴定后去烘干所得残渣质量/g;为皂化加入的NaOH标准溶液体积/mL;为NaOH标准溶液浓度/(mol/L);为样品淀粉滴定消耗HCl溶液体积/mL;为HCl标准溶液浓度/(mol/L)。

式(2)中:DS为淀粉分子上每个葡萄糖单元上的羟基平均被取代的数量。
1.3.3 红外光谱分析
采用KBr压片法。傅里叶变换红外光谱用于采集StNP和StNP乙酸酯样品的红外吸收,识别改性淀粉中存在的特征官能团。取1~2 mg淀粉样品与干燥的KBr粉末混合并压制成薄膜层,然后通过傅里叶变换红外光谱仪进行测定。样本扫描波长范围为4 000~400 cm,平均32 次扫描,分辨率为4 cm。
1.3.4 核磁共振波氢谱分析
取5 mg干燥的淀粉用品,溶解于0.5 mL氘代二甲基亚砜溶剂中,利用核磁共振波谱仪检测核磁共振波氢谱波谱图。测试条件为:60 ℃,30°脉冲角,延迟时间10 s,数据采集时间2 s。
1.3.5 纳米颗粒的形貌表征
用扫描电子显微镜法取一定量干燥的StNPp和不同DS的StNPpAc通过导电胶均匀地分散到样品台上,轻轻吹去多余的浮样,置于真空镀膜仪下喷镀钯金,制成电镜观察样品,在扫描电镜下拍摄具有代表性的不同放大倍数下的淀粉颗粒性形貌,加速电压为10 kV。
1.3.6 粒径和Zeta电位测定
参考文献[22]并适当调整,将纳米淀粉分散在超纯水中,超声处理20 s,配制0.01%的水分散液。将制备好的样品注入粒径测定的塑料比色皿中,放入测量区,等待仪器自动平衡样品,用激光粒度及Zeta电位分析仪测量淀粉颗粒的粒径分布范围。淀粉颗粒和水的折光系数分别为1.54和1.33,测试数据用其平均粒径范围。将上述的淀粉分散液注入Zeta电位测定专用样品池,采用相同的激光粒度及Zeta电位分析仪进行测定。
1.3.7 纳米颗粒的结晶性
采用X射线衍射仪进行分析:Cu(Kα)射线,Ni片滤波,电压40 kV,电流15 mA,扫描范围2为4~30°,扫描速率为8°/min,扫描步长为0.02°,根据衍射峰振动波数、散射峰面积,分析干燥的淀粉样品晶型并计算结晶度。
1.3.8 接触角的测量
移取一定量的StNP乙醇分散液,均匀涂抹在干净的薄玻璃片上,待乙醇挥发完毕,StNP在玻璃片形成一层薄层,将玻璃片放入真空干燥箱中在35 ℃、-0.095 MPa干燥24 h。其后,将玻璃片浸入到装有大豆油的专用正方体比色皿中,置于接触角测试仪操作平台上,用高精度注射器系统控制一滴3 μL的水滴缓慢滴加到StNP薄层表面,在滴加后5 s,5、10、25、30 min对液滴进行拍照,测量其相应的接触角值。接触角采用仪器自带的手动法测量,每个样品的不同位置测量5 次,取其平均值。
1.3.9 纳米颗粒在油水相中的分散
将StNP和StNPpAc分散在1∶1的水与氯仿中,轻轻摇振混匀,静置后,观察其分散情况。按照同样的操作,水与大豆油1∶1,验证纳米颗粒在油水相中的分散效果。
1.3.10 酶解消化与动力学分析
参照文献[23-24]的方法测定体外酶解消化特性。将100 mg样品与10 mL醋酸-醋酸钠缓冲液(pH 5.2,0.1 mol/L)混合摇匀,置于37 ℃恒温箱中预热10 min。然后加入5 mL混合酶液(290 U/mL-淀粉酶和15 U/mL淀粉葡萄糖苷酶),在37 ℃恒温箱孵育酶解数小时(摇床速率200 r/min),前2 h每隔20 min从中吸取0.5 mL酶解液置于刻度试管,立即放入沸水浴中灭活,而后在6 000 r/min离心5 min。用3,5-二硝基水杨酸法测定其葡萄糖含量。用干燥原淀粉颗粒(dry starch granules,DStG)和糊化原淀粉(gelatinized starch,GSt)按上述方法做对照实验。快消化性淀粉(rapidly digestible starch,RDS)、慢消化淀粉(slowly digestible starch,SDS)、抗性淀粉(resistant starch,RS)含量按式(3)~(5)计算:

式中:为酶解前的葡萄糖质量/mg;为酶解20 min葡萄糖质量/mg;为酶解120 min葡萄糖质量/mg;为总淀粉样品质量/mg。
1.4 数据处理
实验均进行3 次重复取均值,采用Excel 2010对实验数据进行统计、计算和绘表,采用Origin 2008对实验参数和结果绘图。
2 结果与分析
2.1 酯化StNP的表征
2.1.1 X射线衍射分析
如图1所示,原淀粉颗粒呈现典型的衍射图谱,在15°~25°处有15°、17°、18°和23°四个强峰,因此原淀粉是一种A型结晶淀粉。StNP和StNPpAc没有出现显著上述形态的峰值,而是表现为一种无定型结晶态。糊化和醇沉使结晶形态发生了变化。完全糊化的原淀粉分子链之间的氢键被水分子破坏,充分伸展,以分子形态存在,结晶形态被破坏。糊化液在超声的空化效应作用下,淀粉链的一部分结构被破坏,发生断裂,分子质量降低,溶液黏度降低。在醇沉中,分子质量降低后的淀粉链在乙醇中不溶解而重排析出,得到纳米颗粒,但由于淀粉链中大量暴露的羟基与水分子相互作用,即使剧烈搅拌也不能完全将这些水分夺走,使醇沉形成的纳米颗粒难以回生紧密的结晶形态,而是形成一种较为松散的无定型聚集状态。这种结晶变化与淀粉纳米晶的颗粒形态有明显区别,因为纳米晶在制备中并没有发生糖链舒展和重排,而仅是原淀粉颗粒发生了自外到内逐层断裂脱除而得。由图1还可看出,乙酰化也无助于StNP结晶形态的恢复。事实上,文献报道的淀粉纳米晶的高度乙酰化也会破坏淀粉的晶体结构,从而形成无定形宽峰。纳米颗粒的这种无定型聚集态,反映了糖链之间聚集并不特别紧密,表面有一定松散度。这种松散的颗粒结构使表面的活性基团得以暴露更多,在理论上有利于提高酯化活性。

图1 X射线衍射图Fig. 1 XRD patterns of native starch and nanoparticles
2.1.2 红外光谱吸收
如图2所示,在StNP的光谱中,3 372 cm处出现了氢键羟基的宽带。吸收带在2 930 cm左右归因于C—H伸缩振动。1 642 cm是淀粉中吸附水的振动吸收。与StNP相比,不同DS的StNPpAc在1 733 cm处表现出强烈的酯基吸收信号,且随着DS的增加,酯基信号强度增加。同时,在1 417、1 372 cm和1 247 cm处新的吸收带可分别归属于—CH基团不对称形变振动、—CH基团对称形变振动和羰基C=O伸缩振动,这些新吸附峰的出现表明产物StNPpAc是在酯化过程中形成的。在天然和经过改性StNP的光谱中,在1 158、1 080、1 024 cm处都有几个可辨别的吸收峰,这归因于葡萄糖单元的C—O—C键拉伸振动,对于酯化淀粉,通过光谱中1 024 cm和1 730~1 750 cm处峰面积的比率可以粗略估计改性淀粉的酯化DS。在930、858、764、611、578 cm处出现的吸收带归因于整个脱水葡萄糖环和糖苷键的拉伸振动,该区域的吸收是多糖类物质(直链淀粉、支链淀粉、纤维素和淀粉)葡萄糖单元的振动状态的普遍模式。综合上述,红外光谱证实了表面改性,表面的羟基已被乙酰基团部分取代。

图2 不同DS StNPpAc的红外光谱Fig. 2 FTIR spectra of StNPpAc with different degrees of substitution
通常由于淀粉的亲水性、紧密结晶和羟基氢键的存在,淀粉的酯化并不容易,一般需要使用二甲基亚砜、,-二甲基甲酰胺或者吡啶等有机试剂溶解后再进行,即使使用了活性酰化试剂酸酐或酰氯,通常也仍然需要在硫酸、对甲苯磺酸、吡啶或离子液体类催化剂的促进下才能得到DS较高的酯化产物。相关文献报道了一种高DS的乙酸酐酯化淀粉的制备方法(DS达到2.66),在淀粉没有完全溶解的情况下,需要浓硫酸催化才能顺利进行,并且重复实验发现该方法由于在90 ℃反应过程乙酸和生成水的影响,产物会发生一定程度的糊化,在分离纯化时堵塞在滤纸层,说明高DS的产物是在结晶结构被一定程度破坏的基础上获得,反过来也说明淀粉的表面活化处理更利于酯化反应。在水媒介中的乙酰酯化反应,则需要碱性环境才能获得低DS的改性产物(DS为0.14以下)。通过对比实验,在没有催化剂的情况下,原淀粉在同样条件下酯化反应活性非常低,红外光谱没有能够检测到酯基的显著吸收;未经表面技术处理的纳米颗粒由于颗粒尺寸小,羟基暴露较多,具有一定的反应活性,在1 733 cm处出现了一个小峰;采用本课题组的表面处理的技术,有效提高了StNP对乙酸酐的反应活性,而且通过改变常温反应和高温反应时间,改变乙酸酐加入量,可以制备出不同DS的StNPpAc(DS如图2所示)。当料液比为1∶5,反应时间6 h后,DS为0.53,红外吸收峰较为显著,如图3所示。这表明经过表面预处理的StNP反应活性更高,表面改性更容易进行。
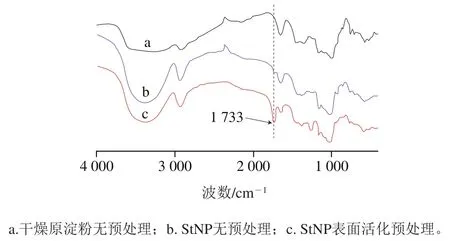
图3 预处理方式不同的淀粉乙酸酯的红外光谱Fig. 3 FTIR spectra of starch acetate subjected to different pretreatments
2.1.3 核磁共振结果
通过核磁共振波氢谱上不同的特征峰,可以判断物质的结构和化学取代基团的区别。淀粉分子上的羟基在酯化反应中被取代。受到取代基团的影响,葡萄糖单元质子的共振会发生变化,取代基上的质子也会发生响应的共振。如图4所示,通过酯化过程,乙酰基被引入到StNP中,由甲基的质子产生的信号出现在1.8~2.2。由于DS不够高,仍然可以观察到脱水葡萄糖单元的在3.29~3.65范围内的特征信号峰,这是因为仅有一部分羟基被取代修饰,糖链骨架上大量羟基依然存在,与相关文献报道相符。

图4 核磁共振谱Fig. 4 NMR spectra of native starch, StNP and StNPpAc
2.1.4 颗粒形貌、粒径及电位分析
如图5所示,原淀粉颗粒呈圆形和椭圆形,颗粒大小在3~20 μm之间。StNP的粒径则远小于原淀粉的粒径,呈球形外观,粒径在30~500 nm范围,这与粒径分析结果相符(图6),其平均粒径为(329.6±23.66)nm,多分散指数(polydispersed index,PDI)为0.456±0.015,颗粒之间有较多桥状黏连,主要是由于淀粉的氢键较强,颗粒之间相互吸引所致,这种作用也是原淀粉颗粒形成较大尺寸的原因。乙酰化处理的StNP表面形貌和边缘略为粗糙,失去了光滑的表面结构和形状,轮廓不清晰,可能是酯化反应和温度影响所致,改性后,颗粒之间存在一定程度的黏连,可能是酯化反应使颗粒表连接大量的乙酰基,基团之间相互作用,这种现象与文献[23]报道使用原淀粉乙酰酯化后在进行纳米化所得的颗粒形貌出现少量黏连相似,但粒径分析表明纳米颗粒在改性前后,尺寸没有发生较大改变(平均粒径(273.8±26.41)nm,PDI 0.412±0.07),这种黏连并不牢固,结构上不紧密,因而并不影响StNP疏水性能在改性得到改善。另外,图6的粒径分布中均有2个大峰,可能的原因是淀粉在糊化和超声破碎时产生了2种分子质量较为集中分布的淀粉链,在醇沉时形成了粒径分布有差异的纳米淀粉颗粒。图6也显示了2种粒径峰的位置在改性前后没有较大变化,说明改性对纳米淀粉颗粒尺寸没有造成显著损害。

图5 原淀粉的扫描电子显微镜图Fig. 5 SEM image of native starch

图6 StNP和StNPpAc法扫描电子显微镜图及粒径分布Fig. 6 SEM images and particle size distribution of StNP and StNPpAc
对于颗粒在水中的聚集状态,Zeta电位分析能解释其中原因,这是了解StNP疏水亲水性能的一种手段。如图7所示,未改性的StNP电位为-12.2 mV,说明其带有一定的负电荷,这与相关文献相符;其负电性可能是由于纳米淀粉颗粒对源于原淀粉本身的微量盐带来的负离子在颗粒表面的吸附所致,另一个可能的原因是超声分散处理时空化效应引起了纳米颗粒表面羟基的微弱解离。经过改性的StNP在纯水中的Ztea电位比未改性颗粒有一定增大,并随着DS升高而增大,可以达到-19.4 mV。在理论上,Zeta电位是表征分散系稳定性的重要指标,在一定程度上反映Stern双电层理论中分散颗粒表面携带同种电荷的数量和粒子之间相互排斥或吸引力的强度,Zeta电位的绝对值越大,该分散体系就会越稳定,不容易发生聚集。因此,在改性后,StNP电位增大,对水的稳定性得到了提高,在水中分散后聚集现象减弱。在文献中,酯化StNP表面电荷因静电斥力能在水环境保持分散状态,这种能力是纳米颗粒能维持乳液稳定性的重要内在动力,对乳液制备等领域起着至关重要的作用。
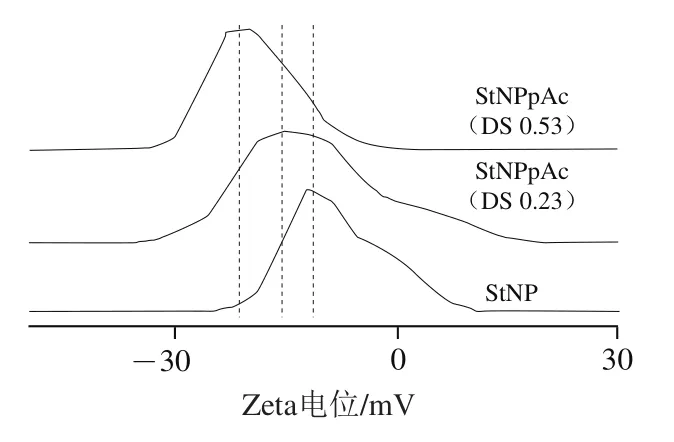
图7 Zeta电位分析Fig. 7 Zeta potential analysis
2.2 疏水性能测试
2.2.1 颗粒表面接触角
如图8所示,未改性的StNP表面接触角值最小,因为StNP的表面富含羟基基团,最有能力与水建立氢键,利于水在表面的润湿。改性后的StNP接触角达到了(109±3.56)°,表明化学处理引起StNP表面极性的剧烈变化,使之具备了一定疏水能力,由此可知乙酰化表面改性改善了淀粉的疏水性能。这种疏水能力随着DS增大而获得增强。如图9所示,不同DS StNPpAc膜上在水滴滴下后接触角随时间的变化情况,淀粉基材的接触角值会随时间而降低,这种现象是由于液滴的在空隙中扩散和渗透所致;接触角值随时间延长而降低的幅度与淀粉的DS有一定相关性,DS高的样品的接触角降低幅度较小,能维持稳定接触状态的时间较长,这说明高DS的表面改性处理能提高淀粉材料表面的疏水性能和稳定性。

图8 不同DS StNP膜上在滴下水滴5 min后的接触角Fig. 8 Contact angle for starch nanoparticle films with different DS values at five minutes after dropping water on them
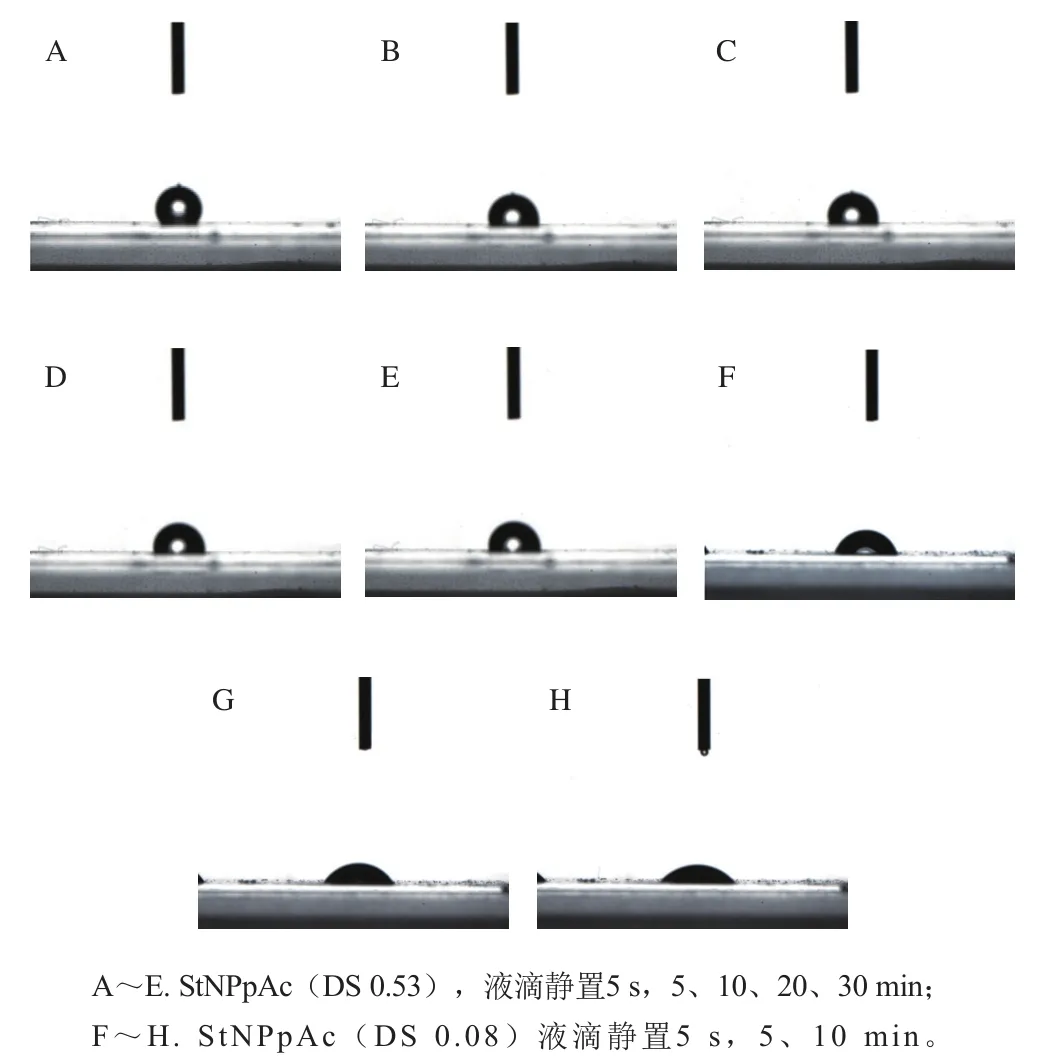
图9 不同DS StNPpAc膜上在水滴滴下后接触角随时间的变化情况Fig. 9 Change in contact angle for StNPpAc films with different DS values changes over time after dropping water on them
2.2.2 润湿性能
疏水性能也可以通过油水混合进行直观表现。将StNP和StNPpAc分散在1∶1的水与氯仿中,静置后,未改性的StNP不能在氯仿中分散,被水层所吸附,改性后,StNPpAc进入氯仿层,并且没有高速均质的作用,也能与氯仿形成微乳,将少量水包裹其中(图10)。在大豆油的分散情况与此类似,未改性的StNP也不容易在大豆油中分散,StNPpAc则与大豆油形成乳液。这说明改性后,StNPpAc更亲油性,具有较好的疏水性能,能对油滴进行包裹,具备了油水乳化的潜力。可见,表面乙酰化修饰改变了淀粉颗粒的亲疏水性能。

图10 StNP和StNPpAc在氯仿/水、大豆油/水中分散情况Fig. 10 Dispersion of StNP and StNPpAc in chloroform/water and soybean oil/water
2.3 酶解消化特性
如表1所示,干燥的未经糊化的原淀粉由于结晶紧密,不容易被酶解,大部分原淀粉颗粒呈抗性特性。StNP和StNPpAc酶解残余量低于65%,表现出更容易被消化的特性,可能因为StNP和StNPpAc是原淀粉经过高温糊化和超声破碎处理,糖链结构不再具有紧密的晶型形态,糖苷更容易被酶接触而水解,可见通过此法制备的StNP和StNPpAc具有较好的生物降解性能和可消化特性。同时,与StNP相比,StNPpAc对淀粉酶敏感性降低,呈现出较强的抗酶解性能,DS高的StNPpAc抗性更强,可能是由于表面乙酰化使颗粒疏水性增强,减弱了酶的界面接触,表现了更好的酶解抗性。当酯化纳米颗粒用于药物、食用乳液制备时,这种相对较强的抗酶解性能使其在体内具有一定的限期稳定性,也使其作为食用多糖应用,具有一定的升糖控制性能。

表1 淀粉和纳米颗粒体外消化特性Table 1 In vitro digestibility of starch and nanoparticles%
从酶解曲线(图11)可见,DStG的酶解率非常低,而GSt酶解率达到79.63%,淀粉糊化后容易引起餐后血糖水平的突然升高。糊化后制备得到的StNP与糊化的原淀粉酶解速率接近,这与StNP在回生中没有形成紧密的定型结晶有关,其松散的无定型结构和极强的亲水性使酶解易于进行。改性得到的StNPpAc,酶解率降低,DS高的产物酶解速率下降明显,这与相关文献相似。特别明显地,较高DS的StNPpAc比StNP酶解率低得多,并且在40~420 min之间仍然具有一定的酶解速率,能持慢速续释放糖分,有利于维持体内的糖分需求。

图11 原淀粉和StNP的酶解曲线Fig. 11 Enzymatic hydrolysis curves of native starch and starch nanoparticles
3 结 论
通过超声破碎处理和醇沉等方式制备了平均尺寸(273.8±26.41)nm的StNP。在不添加催化剂的条件下,StNP的表面乙酰化DS能达到0.53,与未经处理的原淀粉和StNP相比,表面预处理可以较好地提高StNP的表面修饰反应活性。颗粒表面经疏水性修饰,增强了StNP的疏水性能,三相接触角达到了(109±3.56)°,为StNP的后续疏水性领域的应用提供了便捷的技术支持。同时,酯化改性提高了醇沉纳米颗粒的酶解抗性,使其具备一定的控糖性能,为StNP在有血糖要求的食用领域提供更多可能性。
