KLF1基因变异致遗传性溶血性贫血1例
2022-06-06卢琳胡继芬陈丽红何丽丹吴秀燕李建华
卢琳, 胡继芬, 陈丽红, 何丽丹, 吴秀燕, 李建华
遗传性溶血性贫血(hereditary hemolytic anemia, HHA)是由于编码红细胞膜蛋白、红细胞代谢酶及血红蛋白的基因突变引起的一组遗传异质性贫血[1]。HHA病因复杂多样,不同病因引起的溶血常表现出相似的临床特点,如黄疸、肝脾肿大、红细胞寿命缩短或破坏增多、骨髓红系增生、输血依赖性溶血性贫血等,常需进行多项检测才能明确溶血原因,有时甚至存在病因未明现象。目前,已发现的KLF1基因变异可产生广泛的临床表型,如遗传性胎儿血红蛋白持续表达(hereditary persistence of fetal hemoglobin, HPFH)、临界性HbA2水平升高、Lutheran血型、HHA和先天性红细胞生成不良性贫血Ⅳ型(congenital dyserythropoietic anemia Ⅳ, CDA-Ⅳ)等。
KLF1基因位于染色体19p13.13,跨越2.78 kb的基因组DNA,由3个外显子组成,编码362个氨基酸。KLF1蛋白包含2个N端反转录激活域(TAD1、TAD2)、3个C端锌指(ZF1、ZF2和ZF3)形成DNA结合域,能够与相应的靶基因位点特异性结合(图1)。KLF1基因变异所致的HHA呈常染色体隐性遗传,患者临床上常表现为小细胞低色素性贫血、黄疸、肝脾肿大,目前最有效的治疗手段是定期输血,脾切除并不能使该病得到有效缓解,骨髓移植的有效性也尚未见文献报道。此类病例十分罕见,在高通量测序技术广泛开展前,可能存在漏诊或误诊,因此真实发病率可能被低估。
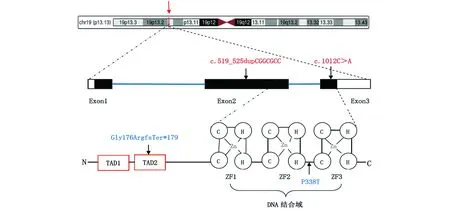
KLF1基因位于19p13,定位用红色箭头表示,由3个外显子组成(Exon1、Exon2、Exon3)。黑框:编码区;白框:非编码区;蓝线为内含子区域;红框为氮端的反式激活域。与本研究突变相对应的核苷酸改变用红色字体表示,与本研究相对应的编码序列改变用蓝色字体表示。KLF1蛋白包含2个氮端反转录激活域和3个碳端(C2H2)锌指结合域。图1 KLF1基因位置及结构图Fig.1 KLF1 gene location and structure
本研究分析1例HHA患儿的致病原因,采用高通量测序技术进行相关基因变异分析,对可疑致病变异位点进行Sanger测序验证,检测到患儿KLF1基因存在c.519_525dupCGGCGCC(p.Gly176ArgfsTer*179)和c.1012C>A(p.Pro338Thr)复合杂合变异,并复习文献发现,该病例为目前已知的KLF1基因突变导致HHA的第6个报道(共累及14例),且在国内为首次报道,现将该病例及文献复习情况汇总如下。
1 病例介绍
1.1 临床资料 患儿,男,5岁,系第1胎第1产,孕母定期产检未见异常,足月顺产,出生时Apgar评分10-10-10,外观无畸形,出生后当天即出现皮肤明显黄染,转入当地医院新生儿科治疗。出生后半个月因皮肤及巩膜黄染加重、持续性高胆红素血症及严重贫血予输血治疗后症状好转,之后每3个月输血1次,维持至今。家族史:父母非近亲婚配且身体健康,否认家族遗传病史。查体:神志清楚,对答配合,中度贫血面容,皮肤、眼睑膜、口唇黏膜及甲床均苍白,全身浅表淋巴结未触及肿大,心肺听诊无明显异常。腹软,无压痛、反跳痛。肝脏肋缘下未触及,脾脏肋缘下2 cm。辅助检查:血常规示白细胞(white blood cell, WBC)15.26×109L-1,血红蛋白74 g/L,平均红细胞体积(mean red blood cell volume, MCV)81 fl,平均红细胞血红蛋白量(mean erythrocyte hemoglobin, MCH)32.5 pg,血小板(platelet,PLT)228×109L-1;外周血检查提示为小细胞低色素性贫血、网织红细胞比例增多;肝功能示丙氨酸氨基转移酶和天门冬氨酸氨基转移酶均正常,总胆红素、直接胆红素和间接胆红素分别为82.0、12.5和69.5 μmol/L。骨髓穿刺可见红细胞系大量增生、核碎裂现象。未检出常见的α、β地中海贫血基因缺失或变异。当地医院进行了血红蛋白电泳、Coombs试验、红细胞渗透脆性试验、葡萄糖-6-磷酸脱氢酶(glucose-6-phosphate dehydrogenase,G-6-PD)试验等相关检查,结果排除了常见的溶血性疾病,如地中海贫血、同种免疫性溶血、遗传性球形细胞增多症、G-6-PD缺乏症等。患儿的生长及智力发育在正常水平。临床诊断“贫血原因待查”。为进一步明确诊断及要求优生咨询就诊笔者医院。本研究经医院伦理委员会审查批准(〔2016〕005号)。
1.2 方法
1.2.1 样本采集及DNA提取 在充分沟通并签署知情同意书后,抽取患儿及其父母外周血各3 mL, EDTA抗凝。采用DNA Mini Kit试剂盒(德国Qiagen公司)提取外周血基因组DNA,-20 ℃保存。
1.2.2 高通量测序检测及Sanger测序验证 将基因组DNA打断成片段并构建基因组文库,通过探针杂交捕获与遗传病相关的基因外显子及邻近内含子区域(±10 bp)进行测序。富集后的DNA片段在高通量测序仪(NovaSeq 6000,美国Illumina公司)上测序。根据与临床表型相关的疑似致病位点设计引物,并对患儿及其父母的目标序列进行Sanger 测序,分析测序结果。
1.2.3 变异位点致病性分析 测序获得的原始数据应用Burrow-Wheeler Aligner(BWA,version 0.7.12)软件进行人类基因组参考序列比对,用Genome Analysis Toolkit(GATK, version 3.3)软件对核苷酸多态性、插入、缺失进行检测和统计分析。用Annovar软件进行变异的筛选和注释,用千人基因组数据库和dbSNP等数据库对检出的变异进行严格过滤,用Mutation taster等软件对可疑变异进行预测分析。严格按照美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics, ACMG)遗传变异分类标准与指南对变异位点进行致病性分析[2]。
1.2.4 文献来源与检索方法 检索以“KLF1”“溶血性贫血”“Kruppel-Like Factor 1”“congenital hemolytic anemia”“hypochromic anemia”为关键词在PubMed、中国知网、中国生物医学文献数据库、万方医学网、维普中文生物医学期刊等数据库进行检索。
1.3 结果
1.3.1 基因测序结果 全外显子组测序发现,患儿携带KLF1基因(NM_006563.3)复合杂合变异,包括第2个外显子c.519_525dupCGGCGCC(p.Gly176ArgfsTer*179)的移码突变和第3个外显子c.1012C>A(p.Pro338Thr)的错义突变,变异分别来源于患儿母亲和父亲。Sanger测序图见图2。
1.3.2 变异位点致病性分析结果KLF1基因c.519_525dupCGGCGCC(p.Gly176ArgfsTer*179)为无功能性变异。根据ACMG指南的判断标准,该变异被判定为致病性变异(PVS1+PS4+PM2)。c.1012C>A(p.Pro338Thr)根据ACMG指南的判断标准被判定为可能致病性(PS4+PM2+PM5+PP3)。
1.3.3 文献回顾分析结果 关于HHA的病例最早于2014年报道,共检索到符合KLF1变异导致HHA诊断的文献5篇,均为外文文献,累及患儿13例,男女比例为10∶2(其中1例文献未诉性别),就诊年龄大多在胎儿期到4岁之间(1例12岁)。病因均为KLF1上的2个核苷酸位点发生复合杂合变异,临床表型极其相似,如贫血、黄疸、肝脾肿大,主要治疗手段均为定期输血。
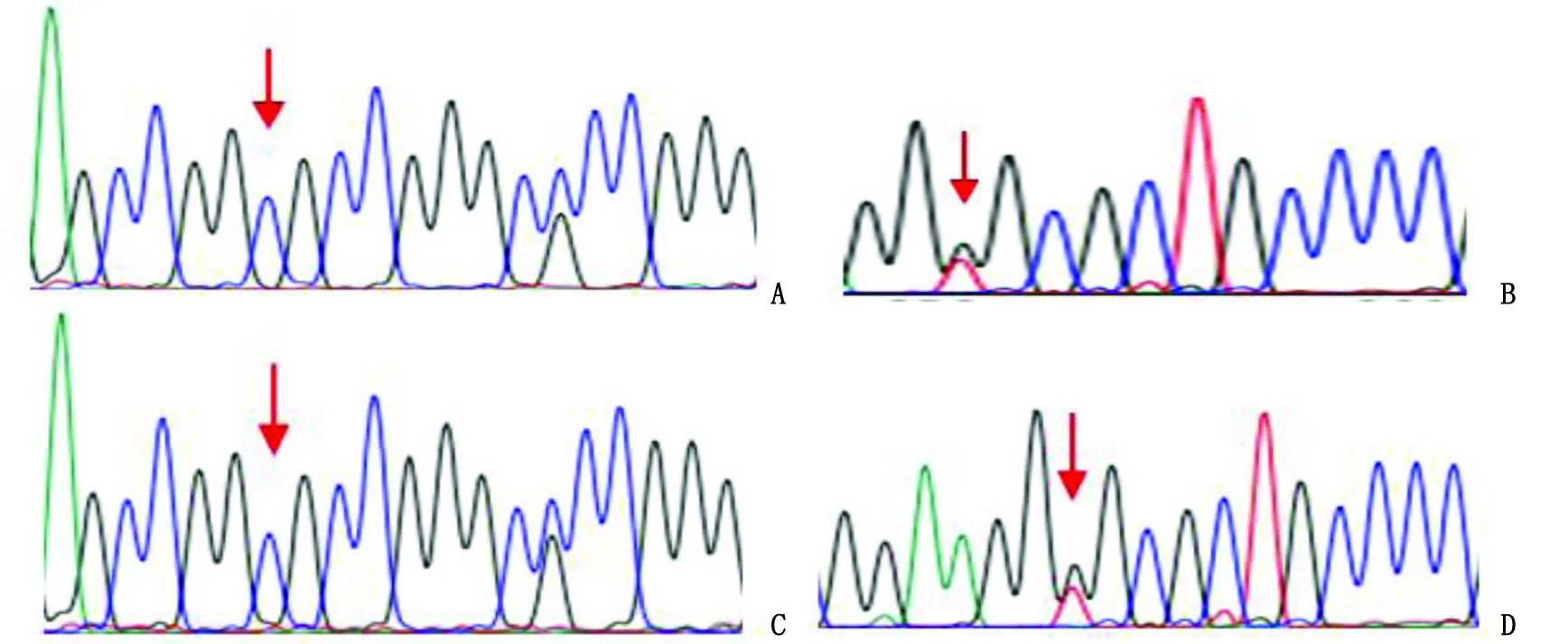
A:先证者c.519_525dupCGGCGCC杂合变异;B:先证者c.1012C>A杂合变异;C:母亲c.519_525dupCGGCGCC杂合变异;D:父亲c.1012C>A杂合变异(红色箭头表示突变位点)。图2 KLF1基因Sanger测序图Fig.2 Sanger sequencing of KLF1 gene
2 讨 论
HHA常发生于婴幼儿期,轻型患者也可能到成年后才被发现。该病是因红细胞的内在先天性缺陷所导致,常见的临床表现有黄疸、肝脾肿大、贫血、高胆红素血症、骨髓造血增生活跃等。治疗手段也因病因不同不尽相同。近年来,HHA的诊疗技术虽有所进展,但仍不尽人意,血库技术的发展大大改善了患儿的预后[3]。
KLF1是一种红细胞生成的关键性转录因子,参与激活、抑制、调节多个血型抗原表达、珠蛋白基因表达和转换、细胞循环及周期、血红素合成酶、细胞膜及细胞骨架、自噬调节、血红蛋白合成等多个环节,几乎控制了红细胞发育与成熟的所有方面[4]。一项针对KLF1基因敲除的小鼠胚胎因严重红细胞缺陷导致死亡的研究也证实,KLF1基因对红细胞的生成起着关键性作用[5]。
KLF1基因突变分为4种类型,主要通过影响蛋白编码序列影响其转录活性[6]:(1)突变位于DNA结构域外,不影响或轻微影响功能。(2)突变在DNA结构域内,包括一些错义突变和小的编码区缺失,能影响基因功能。(3)无义突变或移码突变,能产生截短蛋白,为功能性突变。(4)显性突变,使ZF2上一个高度保守的氨基酸发生错义突变(p.E325K),导致严重的CDA-Ⅳ(#613673)。目前,HGMD数据库收录的KLF1变异已有60余种,尽管锌指结构仅占KLF1的20%,但大部分错义突变均位于这个结构域内或之间[7]。
不同类型的突变可对下游靶基因产生不同的影响,从而引起广泛的临床表型,由表型轻微的罕见In(Lu)(inhibitor of Lutheran)血型携带、HPFH、临界性HbA2水平升高,到严重的遗传性血红蛋白疾病,如CDA-Ⅳ型、HHA,甚至出现胎儿水肿及胚胎致死[8-15]。因KLF1杂合突变常表现为单倍剂量不足[16],大多数携带者通过传统筛查难以发现。在中国南方,KLF1基因突变的携带率为1.25%,而北方携带率为0.08%[17]。但在应用高通量测序技术之前,KLF1基因变异极其罕见。因此由其导致的遗传性血液疾病的病因无法阐明,推测部分死胎可能与该基因功能受损有关。
本例中KLF1基因c.519_525dupCGGCGCC(p.Gly176ArgfsTer*179)为移码变异,会导致所编码蛋白质自第176位甘氨酸开始发生移码,从而产生1个缺失ZF结构域的截短蛋白,为无功能性变异。c.1012C>A(p.Pro338Thr)位于ZF2和ZF3结构域之间,在GnomAD基因组聚合数据库中有收录,在ESP6500siv2_ALL、千人基因组(1000g2015aug_ALL)和dbSNP147等数据库正常对照人群中未发现,该变异在人群中发生频率极低(0.000 05),但在同一个突变位点导致另一个氨基酸发生的变异chr19:12995776G>A(Pro338Ser)已被确认是致病性的,且该病表型与患者症状高度相符。对不同哺乳动物的KLF1进行比对后发现,338位点的Pro保守。经Mutation taster、Mutation Assessor等软件进行预测分析其均有致病可能,会使所编码蛋白质第338位氨基酸由脯氨酸变为苏氨酸。对同源性基因的建模发现,第338位的脯氨酸变为苏氨酸能通过破坏R337和A347之间的氢键,导致二级结构松弛,从而改变ZF1和ZF2之间正常的桥接结构,对KLF功能造成有害影响[18]。
VIPRAKASIT等[13]对8例严重的溶血性贫血患者的研究发现,患儿均是KLF1复合杂合子突变,表型与遗传性非球形细胞溶血性贫血极其相似。总结全球已报道的13例KLF1基因复合杂合变异所致的HHA发现,患儿均携带2种不同的二类突变或是1种二类突变与1种三类突变共存的复合杂合状态;几乎所有患儿都在婴幼儿期发病,临床表现为贫血、黄疸、脾肿大,大部分需要定期输血维持治疗(仅1例在不输血的情况下血红蛋白浓度能维持在80~90 g/L)。本研究中的患儿与文献报道的2例的复合杂合变异完全相同,1例为水肿胎,分别于孕27、29、34周行胎儿宫内输血治疗后,于孕34周分娩,出生后仍需每月输血1次,患儿体质量明显低于同龄儿水平[13];另1例为男婴,出生后有严重的贫血,立即予输血治疗,此后定期输血[19]。但需要注意的是,并非所有的KLF1复合杂合突变都有严重的临床表型,与KLF1突变产生的蛋白类型及对靶基因的影响有关。对其中8例溶血性贫血的患者进行红细胞酶活性检测发现,丙酮酸激酶(pyruvate kinase, PK)缺乏,但对编码PK的相关基因进行测序并未发现变异[12]。进一步研究发现,KLF1基因能特异性地与PKLR基因启动子结合,推测KLF1部分类型的突变可能影响PK的功能,从而产生与遗传性非球形细胞溶血性贫血相似的临床表型,表明KLF1还能与其他基因协同作用参与溶血性贫血的发病。
本病例与已报道的病例有着十分相似的临床特点,与前两例相同突变位点病例不同的是,该患儿在孕期并无胎儿水肿,出生后定期输血的时间间隔也较长,且生长发育正常。推测即使具有相同突变位点的复合杂合变异,临床表型的严重程度也可能存在差异,这为今后的遗传咨询提供了指导信息。
综上所述,笔者对1例HHA患儿进行了KLF1基因变异分析,证实了KLF1复合杂合突变为其致病原因。该报道不仅丰富了HHA的基因诊断,也为进一步研究KLF1基因突变与临床表型严重程度的关系提供了新的依据。
