SMN1基因7号外显子纯合缺失致新生儿期发病的脊髓性肌萎缩症一例
2022-04-01梁晓冰敖当林少珠蔡娜莉刘玲
梁晓冰,敖当,林少珠,蔡娜莉,刘玲
广东医科大学附属医院儿童医学中心,广东 湛江 510180
脊髓性肌萎缩症(spinal muscular atrophy,SMA)是目前致死的最常见的常染色体隐性遗传性肌肉疾病之一,好发人群为婴幼儿[1],由突变的运动神经元存活基因1(SMN1)导致,该疾病在新生儿发病率1/10 000~1/6 000,在人群中的突变基因携带率为1/72~1/47,且不同的种族间的基因携带率具有差异性。主要表现为肌无力和肌肉萎缩,具有进行性、对称性、肢体近端为主的特点。SMA根据发病时间及病程分为Ⅰ~Ⅳ型,各个表型均与SMN1基因外显子7的纯合缺失密切相关(占SMA 患者的95%)。由于SMA具有临床表现复杂、基因拷贝数变化大以及在人群中的携带率较高等多种特点,因此在SMA的早期诊断、遗传咨询、预防和治疗中存在较大的困难[2]。现报道我院新生儿科收治的1例SMA患儿,以加深临床医师对该病的了解。
1 病例简介
患儿,女,23日龄,因“四肢无力23 d”于2020年12月就诊于广东医科大学附属医院。G1P1,胎龄40 周,出生体质量2.5 kg。出生后医生即发现患儿四肢无力,可见肌肉轻微收缩,但肢体不能在床上平行移动,痛觉反应减弱。出生史无特殊,否认双亲近亲婚配,否认遗传学病家族史;其母亲无妊娠合并症;双亲均无烟酒史。查体:神志清楚,精神反应可,前囟约1.5 cm×1.5 cm,平软,无特殊面容,双手呈爪形手(图1),双上肢近端肌力约Ⅰ级,远端肌力约Ⅱ级,双下肢近端肌力约Ⅰ级,四肢近端肌肉松弛,肌张力低。原始反射正常。辅助检查:双耳听力通过。肌电图(EMG):左右胫前肌、腓肠肌内侧头放松状态下呈电静息,轻收缩时运动单位时限正常范围内,募集反应减弱,呈混合相。提示左右尺神经、正中神经、腓总神经、胫神经受损。脑电图:各区以5~20 μv,慢波活动为背景,夹杂少许低波幅快波,左右大致对称,未见明显发作波。头颅磁共振未见异常。基因检测:取先证者及其双亲的外周血,进行了家系增强全外显子组全谱检测,并对临床相关的点突变、拷贝数变异和片段缺失进行了解析。检测到先证者存在一个拷贝数变异(图2):SMN1基因第7号外显子纯合缺失,该纯合缺失片段包含SMN1基因,与脊髓性肌萎缩4型(AR)、脊髓性肌萎缩3型(AR)、脊髓性肌萎缩2型(AR)、脊髓性肌萎缩1 型(AR)相关。诊疗及随访:本案例患儿为新生儿期起病,生后即发现四肢肌力、肌张力明显低下。查体四肢肌力1~2 级,肌张力低,腱反射明显减弱,病理征阴性,深、浅感觉减弱,肌电图提示左右正中神经、尺神经、胫神经、腓总神经受损。基因检测发现该患者SMN1 基因外显子7 纯合缺失。根据SMA的诊断标准,该患儿诊断SMN1 型明确。该患儿出院后未予特殊处理,定期随访,患儿四肢无力呈进行性加重,生长发育迟缓,早期可自行吃奶,后期多次因呛奶致吸入性肺炎于当地医院治疗,患儿于2021年3月因呼吸衰竭而死亡。
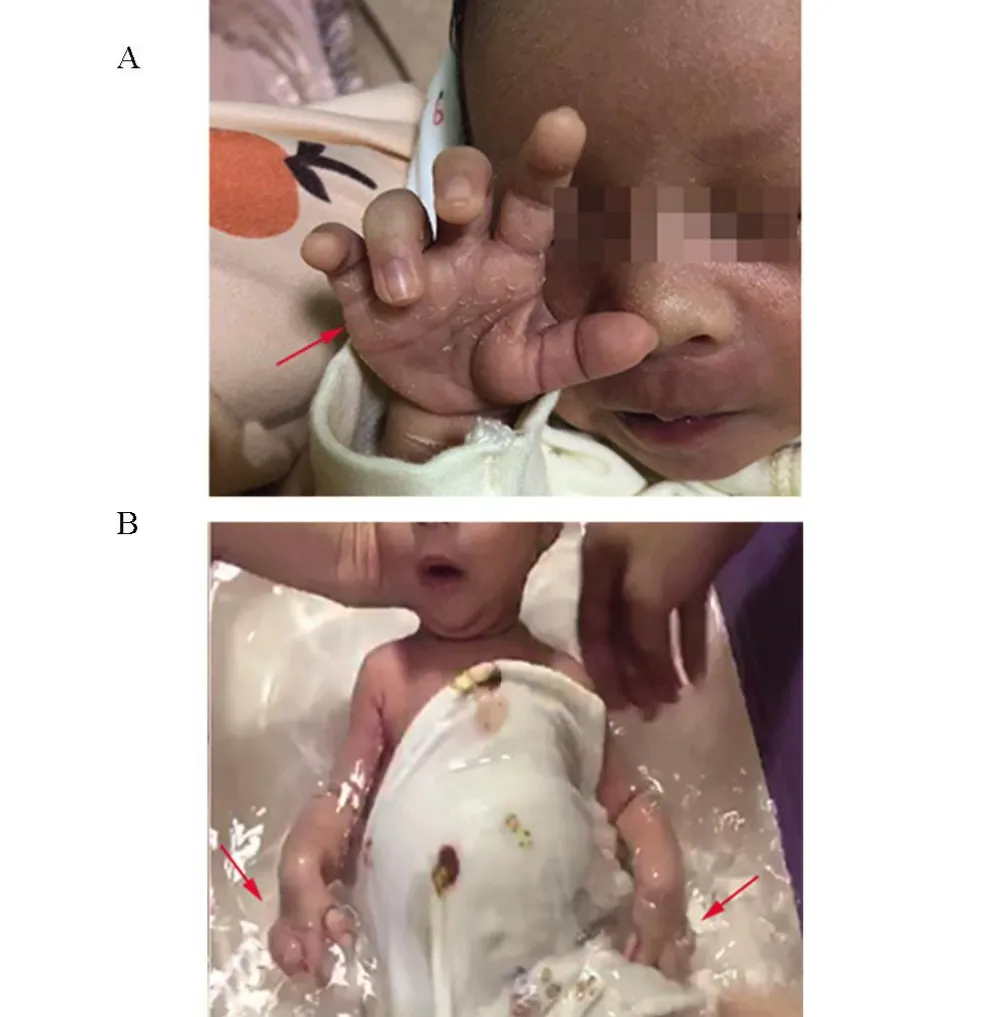
图1 爪形手(双侧,箭头所示)

图2 先证者测序图
2 讨论
该患者无明显家族史,在神经遗传病中,追溯常染色体隐性遗传家族史存在较大困难,其双亲为致病基因携带者可不发病,该患儿为新生儿,遗传图谱难以绘制(因费用问题,该患儿父母未完成家系检测)。脊肌萎缩症是一种常染色体隐性遗传性肌肉疾病,主要由位于染色体5q13.2 上的生存运动神经元基因1(SMN1)突变导致[3]。主要由于脊髓前角细胞退化导致对称性肌萎缩、肌无力;其中,最常见的突变是7 号外显子的纯合缺失[4],导致SMN蛋白缺失[3]、神经细胞萎缩并最终死亡。因相关基因所位于的染色体区域内结构多变,伪基因簇和重复序列较多,容易导致该结构异常,从而增加缺失或转变的次数,故对应的SMN1基因拷贝数组合更为繁杂。其中,SMN1 与SMN2 为两个高度同源基因,前者是致病基因,后者是修饰性基因,SMA患者大部分因SMN1 基因(以第7号和8号外显子为主)纯合缺失所致,少数是因为SMN1基因转化SMN2 基因或SMN1 杂合缺失、点突变所致。SMA根据发病时间及临床表型可分4型:1型、2型、3型及4型,其中前3 型为儿童型。脊髓性肌萎缩症1 型(spinalmuscular atrophy-1;SMA1;OMIM:253300):患者常在生后前几个月内起病,主要临床特征为生长发育迟缓,抬头不稳,四肢及躯体出现严重肌张力减退并呈现肌萎缩的症状,腱反射消失、呼吸费力和吞咽困难等。患者常于2岁前死于肺部感染或呼吸衰竭。脊髓性肌萎缩症 2 型(Spinalmuscularatrophy-2;SMA2;OMIM:253550)常在2岁至成年早期发病,肌电图提示神经源性异常;主要临床表现为患者能独坐,但不能行走;常在青春期时死于呼吸困难。脊髓性肌萎缩症3 型(Spinalmuscularatrophy-3;SMA3;OMIM:253400)通常在18 个月以后发病。肌电图提示慢性病变。主要表现为患者通常可独坐,可独立行走;早期可自己行走,但常常跌倒和行走困难(如爬楼梯),并渐渐失去行走的能力;后期出现脊柱后凸、相关关节劳损,该患者寿命可正常。脊髓性肌萎缩症4 型(Spinalmuscularatrophy-4;SMA4;OMIM:271150)通常在35 岁(20~60 岁)发病;肌电图表现为慢性病变;主要临床表现为部分患者出现小腿肥大,仅存在较小的运动障碍,不累及呼吸系统,该患者寿命正常。
SMA的诊断主要通过临床表现诊断、分子诊断及产前诊断。其中临床表现诊断主要以病例特点、症状体征及辅助检查这3个方面为主:①病例特点,表现为进行性、进行性、以肢体近端为主的肌萎缩与肌无力;②症状体征,肌肉震颤、脊柱后凸等;③辅查,包含实验学检查、神经肌电图、肌肉组织活体检测等。血清肌酸激酶(CPK)检测:SMA Ⅰ型、Ⅱ型均无明显的改变。SMA Ⅲ型CPK 可进一步增高,并随着肌肉的损害程度而进一步加重,后期肌肉萎缩严重时其水平开始出现降低。神经肌电图:提示神经源性广泛性受损。肌活检:大部分肌纤维出现萎缩,呈束性改变,萎缩的肌纤维体积明显减小,在萎缩肌群中可见肥大肌纤维。组化染色提示Ⅰ型、Ⅱ型肌纤维呈簇集改变,两者均萎缩,后者为主。分子诊断为诊断SMA Ⅰ型基因7号外显子缺失的遗传检测首选方法。临床上疑似SMA 或诊断为SMA 的患者均需予基因检测,且患者父母也需进行SMA携带者检测。对于产前诊断,以下均一定是以先证者遗传学诊断已明确为基础:①双亲均为SMA Ⅰ突变的携带者;②一方是SMA患者,另一方是携带者;③双亲的基因结果为携带者与否,以上情况如下次生育时均需予产前诊断。
从脊肌萎缩症的第一次报道开始,目前虽然已有百余年的历史,但治疗效果明显欠佳。其治疗主要包括一般治疗和药物治疗。对于一般治疗,加强护理,尤其是呼吸道的管理,在延缓患儿病情、提高生活质量、延长寿命中扮演着重要角色。大部分患儿死于呼吸道感染,这些患儿常在发病后期由于严重的肌萎缩致无法正常吞咽,容易引起胃食管反流,导致吸入性肺炎。在此期间,需及时予相关的抗生素治疗,加强呼吸道管理,其中预防接种也能较好地阻止肺部感染,必要时可能需要提供一定的呼吸支持(包括机械通气、气管切开等)[5]。康复治疗(如游泳、矫正体态等)也存在一定的疗效。对于药物治疗,在1990 年之前,由于尚未有确定的分子靶向,其临床经验不足,其试验大部分涉及用于缓解疾病症状的药物,部分研试验在以肌无力为主的其他疾病(包括ALS、肌营养不良症)中展现出催人奋进的成果。随着SMA 的遗传机制的逐渐阐明,为应用增加SMN 蛋白表达的方法治疗SMA 提供了分子学理论基础[6-8]。近年来,SMA 的药物治疗取得极大进展[4,8],反义寡核苷酸如Nusinersen 是第一种靶向中枢神经系统的新的基于寡核苷酸的药物[10-11]。当前,诺西那生钠用于鞘内注射治疗SMA,该方法刚起步[12],美国FDA 于2020 年8 月7 日批准基因泰克公司(Genentech,Inc.)新药 Evrysdi(risdiplam)用于 2 个月龄及以上儿童治疗SMA,这是目前FDA 批准的第2 种用于治疗SMA 的药物,也是FDA 批准的第1 种用于治疗SMA 的口服药物。其中,AAV9-SMNI(称为ZolgenSMA)该基因疗法已成为神经肌肉疾病的首个有效疗法,患儿的寿命和运动功能显著提高(直到4年)[8]。但由于这些药物价格昂贵,很多家庭均不能承受,目前仍然以对症治疗为主。该药物的发现为SMA 的药物研发奠定了良好的基础,相信在不久的将来,该病可以被广泛的治疗,造福更多家庭并减轻社会负担[13-14]。
综上所述,SMA 是一种以脊髓前角细胞退化所致的进行性肌肉无力和萎缩为特点、常染色体隐性遗传的神经肌肉性疾病,由于相对罕见,目前依靠临床表现、肌电图检查、肌肉活检、基因检测等手段明确诊断[15],发病率和死亡率较高,尤其是新生儿,其预后极差。因此,产前基因检测很有必要,主要集中在孕10~12 周和孕18~22+6周,分别对绒毛和羊水进行检测[16],其目的是降低该患儿的出生率,减轻家庭痛苦和经济损失,减轻社会重担[17]。为了进一步降低该病的死亡率,目前仍需要更多的临床试验进一步证实相关的药物治疗效果,并希望在不久将来能得到广泛应用。
