COCH基因在遗传性聋及多种相关疾病中的研究进展*
2022-03-28陈永嘉凌捷贺楚峰梅凌云
陈永嘉 凌捷 贺楚峰,3 梅凌云,3
目前已发现与非综合征型聋相关的致病基因有120余个(http://hereditaryhearingloss.org),其中群体凝血因子C同源物(coagulation factor C homology,COCH)基因是首个被报道伴有前庭功能障碍的常染色体显性遗传性非综合征型聋,即DFNA9(deafness autosomal dominant 9)型聋致病基因。迄今为止文献共报道了27个与DFNA9相关的COCH基因突变致病位点,并发现其突变位点与听力和前庭表型密切相关[1~4]。此外,还发现其与梅尼埃病、自身免疫性内耳疾病以及原发开角型青光眼相关联。近年来,基因表达产物cochlin蛋白亚型CTP(cochlin tomoprotein)作为首个诊断外淋巴瘘的金标准也逐步开始应用[5]。COCH基因结构功能、动物模型及致病机制的研究对遗传性聋和多种相关疾病的诊断与防治具有重要意义,但是目前其功能与机制仍不十分明确,故本文对COCH基因在遗传性聋中的最新研究进展及其与多种相关疾病的联系进行综述,以加深对COCH基因型-表型的相关性认识并协助临床相关疾病的诊疗。
1 COCH基因概述
1.1COCH基因结构与结构域 COCH基因(OMIM:603196)定位于14q12-13,包含2 534个碱基,共编码550个氨基酸,具有12个外显子,在多物种内具有高度的保守性,人类与小鼠和鸡的COCH基因氨基酸序列一致性分别达94%和79%(Robertson,1998)。COCH基因所编码的cochlin蛋白是一种分泌蛋白,组成了内耳细胞外基质中非胶原成分的大部分,约占内耳蛋白的70%[6]。
COCH基因结构包含三个重要组成成分:一个短信号肽分子(signal peptide,SP),一个位于N端富含半胱氨酸且与发现于古代无脊椎动物鲎的C因子同源体(factor C homology,FCH)同源的结构域,以及两个位于C端与非胶原结构糖蛋白A型(von-Willebrand factor A,vWFA)同源的结构域vWFA1与vWFA2。FCH与vWFA、vWFA1与vWFA2之间分别被两个中间结构域(intervening domain,ivd)ivd1和ivd2隔开(图1)[7]。
根据三个已鉴定出的包含FCH结构域的蛋白命名,FCH也被称为LCCL(limulus factor C,cochlin,and late gestation lung protein),由第4-6外显子编码。结构预测和循环二色性分析提示LCCL是一个包含α-螺旋和β-折叠链的自动折叠域[8],通过核磁共振光谱学进一步发现LCCL结构域由八个短β-折叠链环绕一个位于中心的α-螺旋构成,并推测该结构内存在空间内相近的四个半胱氨酸所形成的两个二硫键,此区域内发生特定变异的cochlin蛋白将不能折叠成正确的三维结构[9]。此结构域进化上高度保守,在基因编码蛋白的过程中十分重要,目前的研究认为该结构可能与机体免疫系统相关[10,11]。分别由第8-10外显子和第11-12外显子编码的vWFA1和vWFA2存在于多种细胞外基质蛋白中,如软骨基质蛋白、VI型、VII型、XII型和XIV型胶原蛋白等,提示cochlin蛋白可能通过与内耳其他细胞外基质成分结合参与内耳结构的构建(Robertson,1998)。具有vWFA结构域的蛋白质均具有配体结合的特性,通过与胶原纤维或糖蛋白等的结合参与止血、补体系统、免疫系统、剪切力诱导的自凝聚和细胞粘附等(Tuckwell,1999)。
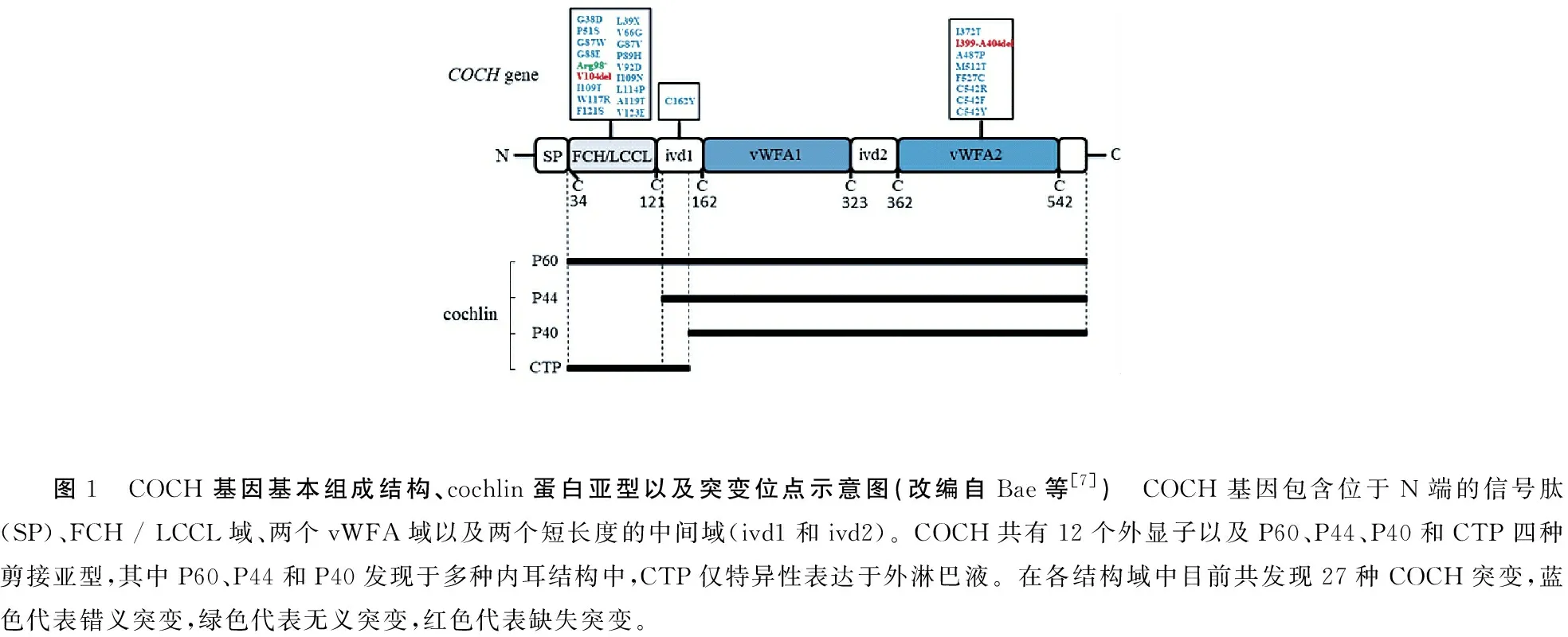
图1 COCH基因基本组成结构、cochlin蛋白亚型以及突变位点示意图(改编自Bae等[7]) COCH基因包含位于N端的信号肽(SP)、FCH / LCCL域、两个vWFA域以及两个短长度的中间域(ivd1和ivd2)。COCH共有12个外显子以及P60、P44、P40和CTP四种剪接亚型,其中P60、P44和P40发现于多种内耳结构中,CTP仅特异性表达于外淋巴液。在各结构域中目前共发现27种COCH突变,蓝色代表错义突变,绿色代表无义突变,红色代表缺失突变。
1.2Cochlin蛋白亚型 目前在人内耳中已经发现有3种糖基化cochlin蛋白亚型,分子量分别为40、46和60 kDa,此外还发现一种特异性表达在内耳外淋巴液中的16 kD小分子蛋白CTP。各亚型的分子大小差距的形成可能与可变mRNA剪接、外显子跳读和翻译后的蛋白水解有关(Robertson,1997)。对出生后天数(day after birth,DAB)不等的大鼠内耳分别进行cochlin蛋白亚型的检测发现早期cochlin蛋白亚型以p63为主,而后p40s和p44s逐渐增加,作者推测p63s对出生后早期耳蜗的发育起着重要作用,而后p40s和p44s可能与促进内耳成熟有关[12]。
1.3COCH基因定位表达研究 COCH基因表达产物在人胚胎与人分布基本一致,在耳蜗和前庭中高表达,而在脑、脊髓、淋巴结、脾、肺等其他组织中表达水平很低(Khetarpal,1991)。COCH基因在成年小鼠中的表达范围则更加广泛,除内耳外,同时在脾中高表达,在大脑、脊髓和胸腺等组织中也有中等水平表达(Robertson,1998)。
2 COCH基因与DFNA9
2.1DFNA9基本临床表型特点 自从1994年Robertson等发现COCH基因以及后来的研究确定其是导致DFNA9的致病基因的这20余年来,已有数十个由COCH基因突变引起遗传性聋家系被报道。DFNA9型耳聋典型表现为20~40岁起病,早期以双侧对称性高频感音神经性听力损失为主,起病20~30年后逐渐发展为全频重度感音神经性聋;患者前庭功能障碍往往与听力障碍同时发生,但前庭表型差异明显,可表现为眩晕、不稳感或向一侧倾倒,也可没有任何前庭功能受损的表现。
2.2DFNA9发病机制的研究 目前已经报道了包含10多个国家和地区的27种与DFNA9相关的COCH突变(表1)。有研究者对其中5种突变类型患者的颞骨组织进行分析发现了类似的病理特点:以螺旋韧带退行性变、伴有非细胞结构嗜酸性物质的沉积为主,同时在远端骨螺旋板、螺旋缘基底层和前庭神经上皮基底的间质组织中也发现同性质物质沉积。定量细胞学分析和免疫标记法证实了骨螺旋缘上的螺旋神经节细胞树突状纤维的退行性变是DFNA9听力损失发生的主要病理因素[13]。
对CochG88E/G88E敲入小鼠模型和Coch-/-敲除小鼠模型的耳蜗和前庭功能研究发现,两种鼠模型都在21月龄时听性脑干反应(auditory brainstem response,ABR)阈值有大幅提升,前者仅表现为高频听力损失,而后者全频均受影响;但Coch-/+小鼠没有听力障碍,CochG88E/+小鼠ABR阈值提升情况与纯合子CochG88E/G88E小鼠相似,因此作者推测单倍体剂量不足不是导致DFNA9听力损失的原因[1,14],而可能是显性负性效应或功能获得性效应。随着近年COCH基因变异相关的常染色体隐性遗传性聋的家系的报导[2,3],功能丢失性效应也可能成为DFNA9致病机制之一。
2.3COCH突变基因位点与听力和前庭表型相关性研究 早期所发现的DFNA9致病变异位点均位于LCCL结构域内,且大多数位于第四和第五外显子[33]。随着报道的DFNA9家系越来越多,vWFA及ivd内也发现了相关致病性突变位点。一项综合了21种已知COCH突变的DFNA9患者信息的基因型-表型相关性研究发现,位于vWFA结构域变异的突变个体主要表现为听力损失,而含有LCCL结构域变异的个体往往还伴有前庭功能障碍[7]。近年来报导的新DFNA9家系也再次证实其基因型-表型的联系[2,3](表1),国内的一项研究发现具有相同类型变异的DFNA9家族也可能具有明显不同的表型,作者分析这可能是由于COCH突变导致了突变体cochlin的LCCL区域片段断裂或聚集而使得两家系表型不同[34]。因此,不同的COCH突变具有不同的病理机制,甚至相同的COCH突变也可能有多种病理机制。未来需要继续扩增COCH变异的遗传学信息资料,丰富不同种族地区DFNA9患者的听力与前庭表型数据库,进一步研究COCH突变的基因型与听力和前庭表型的相关性以及致病机制。
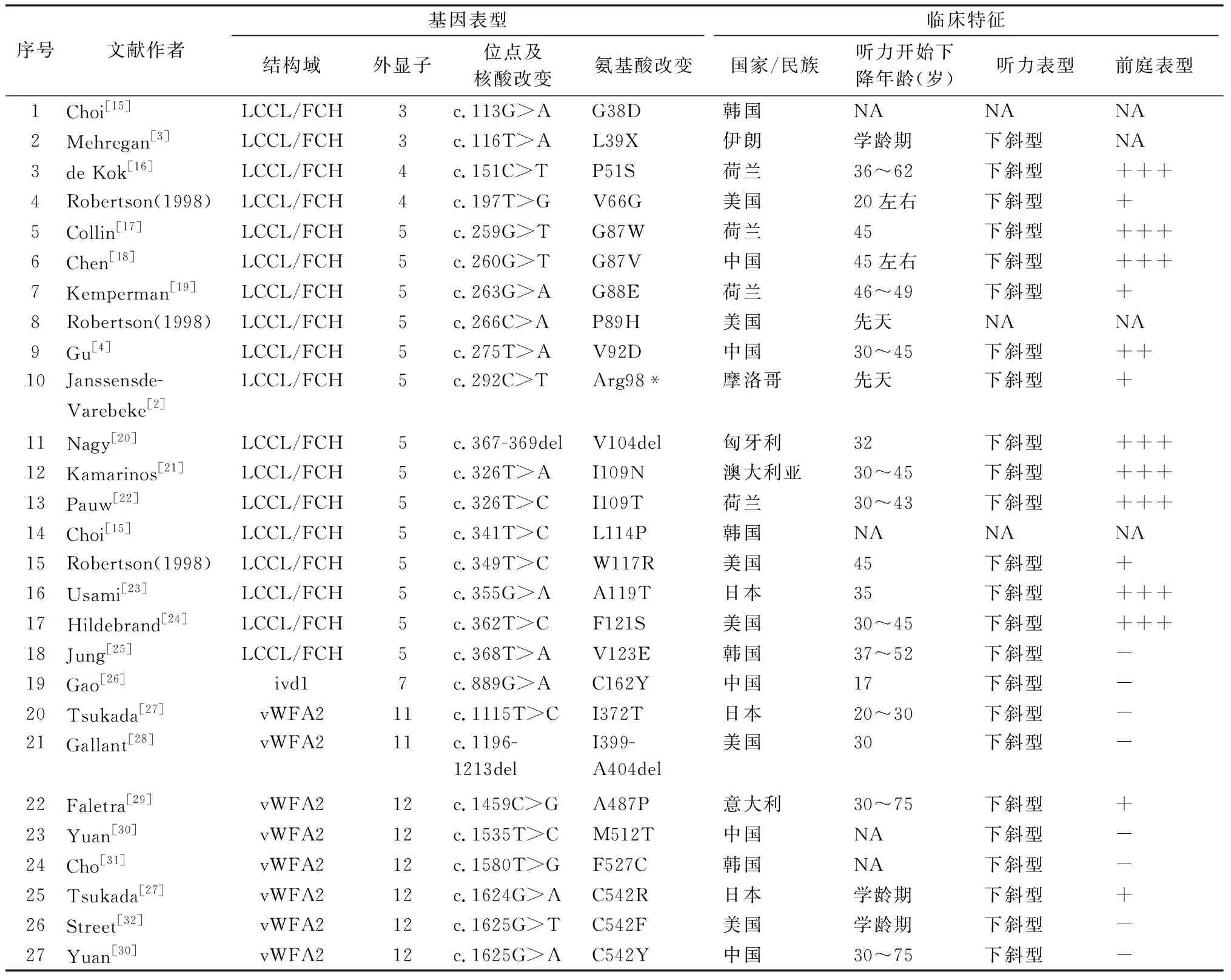
表1 各文献报道的COCH基因表型突变及临床特征
3 COCH基因与其他相关疾病
3.1COCH基因与梅尼埃病 梅尼埃病(Meniere disease,MD)是以发作性眩晕、感音神经性听力损失、耳鸣和/或耳闷胀感为主要症状的一种内耳功能紊乱性疾病,组织病理学改变为膜迷路积水,但目前确切的病因和发病机制仍不十分明确。大多数的梅尼埃病报导为散发性,但在欧洲等梅尼埃病较高发地区发现部分病例具有很强的家族聚集性和遗传早发现象,且种族、人口发病率和地理分布的差异也支持遗传因素在该病发展中的作用。欧洲的两项多中心、横断面研究回顾了1 557例确诊单侧梅尼埃病患者和405例双侧梅尼埃病患者的临床信息,根据MD患者的遗传史、有无偏头痛以及自身免疫性疾病等,通过聚类分析将单侧MD和双侧MD分别定义了5种临床亚型,其中具有梅尼埃病家族史的患者被归为其中重要的一种[35,36]。
目前梅尼埃病基因学研究主要包括COCH、HLA、NFKB、KCNE、MIF等,其中COCH是早期研究较多的一个基因。早期发现的三个DFNA9家系中发现约25%的患者临床表现符合梅尼埃病的诊断标准,随后也有多项研究[37,38]报导了DFNA9患者中有类似梅尼埃病的症状,因此,推测COCH可能是梅尼埃病的致病基因;但后续研究没能证实COCH与梅尼埃病有明确的相关性[39,40],Usami等[23]对23例常染色体显性遗传性听力损失患者(其中4例有前庭症状)和20例梅尼埃病患者进行了COCH基因分析,认为COCH基因突变是造成相当一部分常染色体显性遗传性听力损失患者伴有前庭症状的病因,而与梅尼埃病所表现出来的前庭症状不相关。COCH基因与梅尼埃病的相关性有待进一步验证,临床医师应特别注意梅尼埃病与DFNA9的鉴别[41]。在得益于高通量测序技术的进步的同时,未来也需要收集更多有详细临床信息的大家系来揭示梅尼埃病潜在的遗传致病机制。
3.2Cochlin蛋白与自身免疫性内耳疾病 自身免疫性内耳疾病(autoimmune inner ear disease,AIED)是一种少见的、通常免疫抑制剂治疗有效的内耳疾病,表现为双耳不同程度发作、在3~90天内进行性加重或波动性变化的感音神经性聋,约50%的患者可同时伴有眩晕、耳鸣或耳闷胀感等[42]。由于缺乏特征性的检测手段,目前对AIED的诊断依赖于典型的临床症状、对糖皮质激素的反应阳性以及其他相关常见病的排除,因此很容易被误诊。而早期诊断对于糖皮质激素和免疫抑制剂治疗该疾病的效果尤为关键,故明确致病原因以及找到早期诊断分子标记物尤为重要。
研究发现cochlin特异性的分泌IFN-γ的CD4+和CD8+T细胞可能参与了AIED的病理过程,与正常听力人群以及噪声或年龄相关性听力损失患者对比,AIED患者血浆中可检测到显著增高滴度的特异性cochlin IgG抗体,提示cochlin蛋白可能是调节自身免疫性内耳炎症和听力损失的重要靶点,这为通过酶联免疫斑点检测法检测分泌IFN-γ的T细胞或cochlin抗体滴度协助诊断AIED提供了思路[43,44]。多种曲霉属和青霉属真菌与cochlin蛋白的LCCL域具有同源性,研究发现在这些真菌的刺激下AIED患者血浆中所检测出的抗真菌抗体和抗cochlin IgG抗体比正常人群高,同时外周血单核细胞所产生的免疫细胞因子IL-6和IL-1β水平也增高,因此,作者推测这些与LCCL同源的真菌的暴露参与了AIED的病理过程,可诱发部分AIED易感患者的疾病的发生[10]。Cochlin蛋白和相关抗体为AIED的诊断提供了新的思路和前景。
3.3Cochlin蛋白与外淋巴瘘 外淋巴瘘(perilymphatic fistula,PLF)是由于头部外伤、气压伤、慢性炎症等原因在内耳外淋巴液腔与中耳腔、乳突腔或颅腔之间形成异常沟通产生的,主要表现为突发性、渐进性或波动性感音神经性聋,伴有眩晕或呕吐。如果能早期明确诊断可通过手术治疗避免永久性听力损失,但是由于缺乏金标准,仅仅通过临床表现和影像学很难对外淋巴瘘进行确诊。
有研究通过cochlin蛋白亚型抗体的检测首次在内耳外淋巴液中发现一种仅含有LCCL域的新cochlin亚型CTP,而在脑脊液、唾液和血清中未检测到该种蛋白的表达[45]。研究者收集23例临床诊断的特发性自发外淋巴瘘患者的中耳灌洗液(middle ear lavage,MEL) 样本进行CTP特异性检测,发现其中约有50%病例检测CTP呈阳性,且病例对照研究发现起病7天内手术组术后听力改善优于发病7天后手术组[46]。一项多中心、大样本研究对不同病因造成的497例可疑PLF患者和其他一些内耳相关临床表现患者的MEL进行CTP检测,可以在相当一部分有内耳相关表现而无论有或无前驱事件(如航行、潜水、外伤等)的患者中检测出CTP阳性,这些CTP检测阳性的患者临床表型变异大,因此,可能相当一部分原因不明、具有耳蜗-前庭症状的患者被忽视或误诊[47]。CTP作为一种客观的新应用型的生物标志物可为PLF的诊断提供强大的依据,对CTP的不断探索将为PLF的诊断展现更光明的前景[5]。
3.4COCH基因与原发开角型青光眼 青光眼是不明原因导致视神经节细胞退行性变和不可逆性视力损伤的进展性疾病。原发开角型青光眼(primary open angle glaucoma,POAG)是其最常见的类型,目前对POAG的病理学研究认为cochlin蛋白的沉积与眼内压增高相关。在对POAG患者及DBA/2J小鼠模型研究中发现,随着小鼠年龄的增加,作为调节房水循环十分关键的小梁网(trabecular meshwork,TM)中的cochlin蛋白沉积也随之增加,而Ⅱ型胶原蛋白随之减少,在正常人群及其他非青光眼鼠模型中未发现cochlin沉积[48];因此,作者认为cochlin的沉积可能改变了TM的结构使得房水流出受阻而影响了前房水循环,随着时间进展导致了眼内压病理性增高。
4 总结与展望
综上所述,COCH基因突变可引起伴或不伴眩晕的非综合征型常染色体显性遗传性聋。DFNA9型耳聋具有明显的基因型-表型联系,位于vWFA域突变的患者绝大多数表现为早期高频听力下降、后期全频受累的感音神经性聋,而LCCL域变异的患者往往还伴有不同程度或外显率的前庭功能障碍。高通量测序技术和遗传学的高速发展促进了COCH基因结构与功能、突变与致病机制的研究,在更多耳聋患者和正常人群中对COCH基因进行捕获分析并建立完善的数据库将为DFNA9和其他常染色体显性遗传性聋的诊断提供参考。COCH作为 DFNA9与梅尼埃病、自身免疫性内耳疾病和原发开角型青光眼等疾病间沟通的桥梁,对COCH更深入的研究将有助于更好地理解相关疾病的致病机制以及疾病间的潜在联系,同时也提示在临床诊疗中要对这些疾病进行联系和鉴别。此外,利用CTP的特异性研发高精度、简便快速的金标准诊断试剂盒,将有助于外淋巴瘘的早期诊断和预后评估,并有望通过早期手术治疗减少永久性听力损失和并发症的发生。
