一个非综合征型聋家系MITF基因致病性新突变△
2022-03-28高玲丽潘春晨鲍坚强孙敬武
高玲丽 潘春晨 鲍坚强 孙敬武
耳聋是一种极其复杂的高度遗传异质性疾病,病因主要包括遗传与环境因素,但是以遗传因素为主(60%);根据病变累及范围分为综合征型聋和非综合征型聋。同样的耳聋表型可以由不同基因变异导致,或者同一基因可以导致不同的耳聋相关临床特征[1],同一个基因突变既能引起综合征型聋,也能引起非综合征型聋。迄今为止,已经鉴定出121个非综合征型聋基因,其中有49个与常染色体显性遗传非综合征型聋(ADNSHL)相关[2]。在我国耳聋人群中,最常见的致病基因是GJB2、SLC26A4、mtDNA12SrRNA和GJB3,约占30%~50%[2]。既往报道MITF基因突变会引起Waardenburg综合征和Tietz综合征[3],两种疾病均为常染色体显性遗传性综合征型聋。随着新一代测序技术的发展,越来越多的耳聋基因被发现,近年来研究发现MITF基因突变除了导致综合征型聋,也会引起非综合征型聋[4,5]。本研究发现了一个涉及3代的非综合征型聋家系,通过遗传性耳聋基因芯片、耳聋基因靶向测序以及Sanger测序技术,证实该家系患者中存在MITF基因c.730G>A(p.Gly244Arg,NM_000248)突变,这是中国人群中首次检测出MITF基因该位点突变引起非综合征型聋,报道如下。
1 资料与方法
1.1家系资料搜集 家系调查由中国科学技术大学附属第一医院耳鼻咽喉头颈外科于2019年9月5日搜集完成;家系成员居住地为安徽省,汉族。对家庭成员进行了详细的病史采集和全身体格检查,包括对先证者及家系成员进行完整病史(出生史、耳聋病史、家族史、外伤史、噪声暴露史、耳毒性药物应用史等)采集和系统的全身体格检查。本调查研究获本院伦理委员会认可,所有参与的研究对象均签署了知情同意书,未成年人由监护人签署知情同意书。
1.2听力检测及家系图绘制 对先证者及其家庭成员进行了纯音听阈及听性脑干反应检测。观察先证者及家系患病成员行走姿态,初步排除了共济失调或步态不稳等前庭功能异常的症状;根据病史排除了外伤、药物、噪声暴露等听力损失风险因素,根据家系调查和听力结果绘制系谱图(图1)。

图1 非综合征型聋患者的家系图谱
1.3DNA提取 采集家系中Ⅰ-4、Ⅱ-3、Ⅱ-4、Ⅱ-5、Ⅲ-3、Ⅲ-4成员的外周血各3 ml,EDTA抗凝,用血液基因组DNA提取试剂盒(天根生化科技北京有限公司)提取样本DNA,用Nanodrop2000超微量分光光度计检测DNA的纯度和浓度,取适量样本稀释至浓度100~200 ng/μl,其余-20 ℃保存。
1.4遗传性聋基因检测 应用9项遗传性耳聋基因芯片对该家系中的Ⅰ-4、Ⅱ-3、Ⅱ-4、Ⅱ-5、Ⅲ-3、Ⅲ-4成员进行中国人群中常见的4个耳聋基因9个突变位点GJB2(35delG、176_191del16、235delC、299_300delAT),GJB3(538C>T),线粒体DNA 12SrRNA(1494C>T、1555A>G),SLC26A4(2168A>G、IVS7-2A>G)的检测。
1.5耳聋基因靶向测序 应用已知遗传性聋基因捕获试剂盒,对遗传性聋相关基因编码区及附近剪切区的DNA进行靶向测序,包括非综合征型聋相关基因、综合征型聋相关基因、听神经病相关基因及一些与耳聋相关需鉴别诊断的基因,共415个基因。方法:取3 μg基因组DNA,用1×low TE Buffer稀释到30 ng/μl,用Covaris-S220超声波打断仪将基因组DNA随机打断成约150 bp左右的片段准备构建文库。对这些DNA片段按照Illumina的规定进行末端修复及添加Illumina适配元件。经过PCR扩增,生物素标记的单链DNA捕获探针与文库DNA杂交,磁珠吸附抓取目标基因,洗脱纯化富集。获取的基因片段文库加载到测序芯片Flowcell上,在Illumina NextSeq 500测序仪上对415个耳聋相关基因外显子进行高通量测序,此项工作由北京迈基诺医学检验所完成。
将原始测序数据去除污染和接头序列,然后利用BWA软件将过滤后的序列比对到NCBI数据库人类基因组参考序列(hg19)上,利用GATK软件分析得出单核苷酸变异(single nucleotide variation,SNV)和插入缺失突变(inserts and deletions,INDEL)的相关信息。然后通过ANNOVAR软件对所有的SNP和INDEL进行注释。筛选正常人数据库中频率小于0.05的突变位点,正常人数据库包括千人基因组计划(http://www.1000genomes.or/)、Exome Variant Server(http://evs.gs.washington.edu/EVS/)和EXAC(http://exac.broadinstitute.org/)。错义突变使用SIFT,PolyPhen-2,MutationTaster和GERP++等软件进行致病性预测和保守性预测,剪切位点的改变用SPIDEX软件分析其致病性。
1.6突变验证 利用Sanger测序验证经耳聋基因靶向测序得到的候选基因,进行家系共分离验证,排除其他候选基因,最终锁定致病基因MITF基因。针对检出的致病基因MITF突变位点设计验证引物。MITF扩增的引物如下:正向:5′TGTAACCAAGCACCACCTGT3′;MITF反向:5′CCACTCTGCACATGTCCAAG3′。使用贝克曼自动化工作站预设程序配置PCR扩增反应体系,并按如下步骤运行程序:96 ℃ 1 min 30 s; 96 ℃ 15 s, 50 ℃ 6 s, 60 ℃3 min 30 s,28个循环;4℃ Holds。PCR产物使用3130XL测序仪进行毛细管电泳测序并在ABI 3130 Genetic Analyzer (Applied Biosystems)上进行分析,再在家系成员中进行共分离验证。并选取家系外10例听力正常人,对患者所发现的可疑致病变异位点中进行人群携带率验证。
1.7保守性及蛋白三维结构分析 使用Clustal Omega软件(https://www.ebi.ac.uk/Tools/msa/clustalo)对多个脊椎动物蛋白序列(鸡、小鼠、大鼠、人、狗和牛)进行比对。利用SWISS-MODEL(https://swissmodel.expasy.org)工具进行同源建模模拟MITF蛋白的三维结构,分析该位点突变前后对该蛋白质功能的影响。
2 结果
2.1家系表型及听力检查结果 该家系共3代15人,4人患病,均诊断为先天性双侧感音神经性聋,表现为常染色体显性遗传,家系图见图1。先证者(Ⅲ-4),男,2岁4个月,先天性聋;姐姐(Ⅲ-3)、母亲(Ⅱ-4)、外婆(Ⅰ-4)均为先天性聋;父亲(Ⅱ-3)及其他家属(Ⅰ-1、Ⅰ-2、Ⅱ-2、Ⅱ-5)听力正常。先证者(Ⅲ-4)及姐姐(Ⅲ-3)在90 dB nHL刺激强度下双侧听性脑干反应未引出。先证者母亲(Ⅱ-4)0.25~8 kHz纯音气导平均听阈右耳93、左耳82 dB HL;先证者外婆(Ⅰ-4)右耳68、左耳63 dB HL,均诊断为双耳重度感音神经性聋(表1)。

表1 家系中Ⅰ-4和Ⅱ-4成员各频率纯音听阈(dB HL)
2.2遗传性聋基因芯片检测结果 家系中Ⅰ-4、Ⅱ-3、Ⅱ-4、Ⅱ-5、Ⅲ-3、Ⅲ-4成员的9项遗传性聋基因芯片检测结果均为野生型,未发现中国人群中最常见的耳聋基因位点突变。
2.3耳聋基因靶向测序结果 对先证者Ⅲ-4进行了耳聋基因Panel靶向测序,数据分析结果显示其携带4个基因的一个单杂合变异,包括:ILDR1: c.772C>T(p.Gln258stop)、CLDN14:c.2T>C(p.Met1Thr)、MITF: c.730G>A(p.Gly244Arg)、TNC: c.473G>A(p.Arg158Lys)。
2.4变异确认及家系共分离验证结果 Sanger测序结果证实了上述变异真实存在,对先证者全家四人检测这四个位点的携带情况,发现先证者的3个变异,包括:ILDR1: c.772C>T(p.Gln258stop)、CLDN14: c.2T>C(p.Met1Thr)、TNC: c.473G>A(p.Arg158Lys)来源于其父亲(图2),表明靶向测序所发现的这3个变异均与临床表型不符合共分离现象。变异MITF: c.730G>A(p.Gly244Arg)来源于其母亲,父亲(Ⅱ-3)未发现该位点的变异,为野生型;随后进一步验证Ⅰ-4和Ⅱ-5,Ⅰ-4为突变型,Ⅱ-5为野生型,临床表型符合家系共分离现象(图3);最终锁定MITF为候选致病基因。利用蛋白功能预测软件REVEL进行MITF基因变异致病性的分析,结果显示为D(预测为有害)(表2),提示其可能为一个潜在的致病位点。
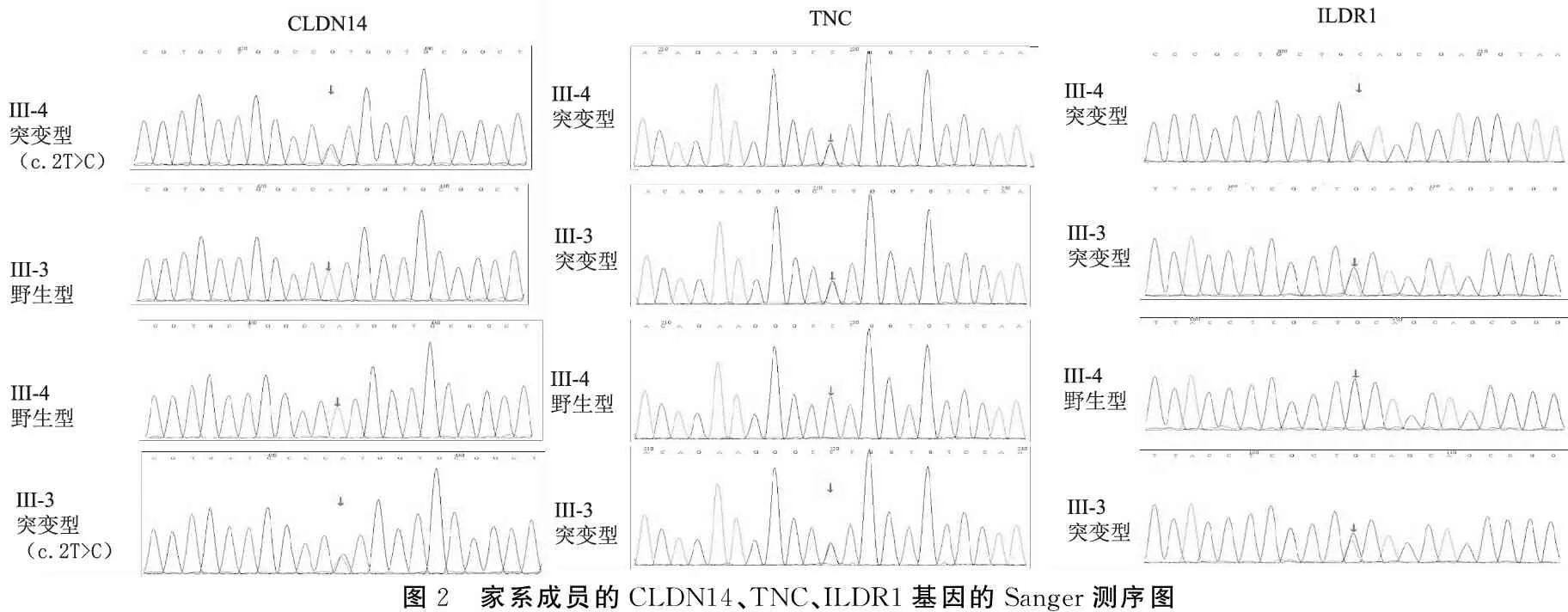
图2 家系成员的CLDN14、TNC、ILDR1基因的Sanger测序图

图3 家系成员MITF基因的Sanger测序图

表2 MITF基因高通量测序结果分析
2.5家系外对照者变异验证结果 选取的家系外10例听力正常人作为对照,用Sanger测序法对先证者所发现的MITF基因变异(c.730G>A)进行筛查,结果均未携带上述变异。
2.6MITF物种保守性分析 使用Clustal Omega软件(https://www.ebi.ac.uk/Tools/msa/clustalo)对多个脊椎动物蛋白序列(鸡、小鼠、大鼠、人、狗和牛)进行比对,结果显示244Gly在各物种中高度保守,该突变体位于MITF的碱性螺旋-环-螺旋(bHLH)区域(MITF-M亚型)中。
2.7MITF蛋白三维结构预测 预测的MITF蛋白三维结构(图4,箭头标注示突变位点)来源于MITF残基226~257,MITF基因是一个编码含bHLH-LZ(basic helix-loop-helix zipper)结构域的转录因子,与DNA结合以二聚体的形式存在,可以看出Gly244位于第二个螺旋的开头,靠近蛋白质-DNA界面,所以该位置的突变很可能增强MITF与非特异性DNA的结合。
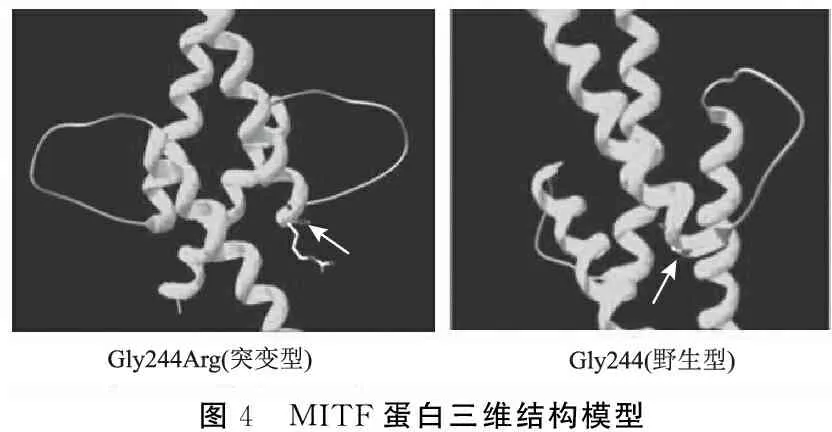
图4 MITF蛋白三维结构模型
3 讨论
MITF基因(melanocyte inducing transcription factor)在各种细胞类型的建立中起关键作用,主要对黑色素细胞的增殖、迁移、存活和分化至关重要,通常又被称为该谱系的主要调控因子[7];它是色素细胞发育的关键基因,通过Myc超基因家族的基本螺旋-环-螺旋-亮氨酸拉链(basic helix-loop-helix leucine zipper; bHLH-Zip)功能结构域发挥作用[8]。在内耳中,MITF基因的功能是通过酪氨酸酶调节黑色素的产生,并直接参与听觉的分子过程,黑色素细胞存在于血管纹层的中间层。这些黑色素细胞(所谓的中间细胞)具有功能性Na+-K+-ATPase,该酶控制维持内淋巴正常体积和离子浓度(循环)所需的离子和物质运输,因此,对于调节血管纹层的完整性和维持正常的听力至关重要。仅内耳的黑素细胞具有此功能,而其他色素组织中的黑色素细胞则不具有这种功能[5]。在小鼠中,中间细胞的耗竭会导致耳蜗内电位大幅降低、内淋巴间隙严重变性、感觉毛细胞崩溃和损失,并伴有明显的听力损失[9]。
MITF/Mitf功能与人类和小鼠的许多遗传性疾病有关[10]。既往文献报道MITF基因缺陷与黑色素瘤[11]、眼白化病[12]、Waardenburg综合征及Tietz综合征[3]相关,其中,Waardenburg综合征及Tietz综合征为综合征型聋。近年来随着研究的不断深入,有学者发现MITF基因突变也会导致非综合征型聋,例如:2018年,在云南-贵州地区发现了一个家系中由于MITF基因杂合突变(c.718C>G, p.Arg240Gly)引起的常染色体显性非综合征型聋[4]。2020年,Supranee thongpradit等发现MITF基因纯合突变(c.1022G>A, p.Arg341His)与人类常染色体隐性非综合征型聋相关[5]。
本家系中3代4人患病,均为先天性双侧感音神经性聋,通过病史采集与咨询,未发现该家系中有皮肤毛发色素改变、虹膜异色、内眦间距异常、上肢异常、巨结肠等,故判定该家系为非综合征型聋家系。本研究首先对中国人群常见4个耳聋基因(GJB2、GJB3、SLC26A4和mtDNA 12SrRNA)9个突变位点进行筛查,排除了常见致病基因位点的突变;随后,通过耳聋基因靶向测序筛选出MITF基因的一个新的突变体,并对该突变进行Sanger测序验证,发现该突变与家系耳聋表型共分离,并且选择家系外的10例正常听力人作为对照,未发现该基因位点突变。本研究检测到的变异位点c.730G>A(p.Gly244Arg, NM_000248)在人群中的携带率极低,且检出者表现为双耳极重度感音神经性聋,经该家系验证分析,先证者及其姐姐、母亲、外婆均为极重度感音神经性聋且均携带该位点杂合变异;先证者之父、舅舅听力正常,该位点无变异,表明该变异来源于先证者母亲。结合家系图可以推测,该家系的遗传模式可能为常染色体显性遗传或线粒体遗传;通过耳聋基因靶向测序未发现线粒体相关基因突变,故可排除线粒体遗传,表明该家系为常染色体显性遗传性聋,这也与测序结果MITF基因发生杂合突变相对应。
本研究发现此变异位点位于bHLH-Zip结构域[13],最早认为此结构域与色素细胞的发育相关,随着研究的不断进展,近年来发现,此结构域不仅在色素细胞的发育中起作用,在耳聋患者中发现的突变位点也位于该结构域[5],表明该结构域与耳聋的发生发展有关,但具体机制尚不明确。Steingrimsson等[14]在小鼠中发现mitf基因Gly244的突变,该位点的突变可能会潜在影响其与DNA的结合,增强与非特异性DNA的结合。经由物种保守性分析,该位点在各物种中高度保守,结合Swiss model预测的蛋白质三维结构图,推测该位点的突变很可能也是通过扰乱MITF与DNA的特异性结合,从而影响MITF基因编码的蛋白质发挥正常功能。该变异的检出将有助于MITF蛋白的功能分析和耳聋的基因诊断。
综上所述,MITF基因c.730G>A(p.Gly244Arg, NM_000248)位点突变可能是本研究中非综合征型聋家系的致病突变,这是首次在中国非综合征型聋人群中检测到该基因位点突变,其是一个致病突变,建议产前做好遗传咨询,降低聋儿的出生率。
