解毒软肝合剂质量控制方法研究
2022-03-23刘丽琴周晓霞陈红斌
刘丽琴 周晓霞 汪 涛 陈红斌 王 莹
解毒软肝合剂来源于浙江省中西医结合医院解毒软肝协定方,有十余年临床使用经验,治疗急慢性肝炎效果显著。解毒软肝合剂由白花蛇舌草、党参、垂盆草、米仁、板蓝根、焦山楂、叶下珠、乌贼骨、木瓜、五味子、土茯苓、炙甘草、赤芍等药材组成。其中白花蛇舌草、叶下珠、土茯苓、板蓝根清热解毒;垂盆草清热利湿;五味子益气养阴;赤芍清热凉血、活血祛瘀;党参、米仁、焦山楂、木瓜和乌贼骨健脾和胃;炙甘草调和诸药,全方共奏清热利湿、健脾和胃之效。该复方未建立完善的质量检测体系以评价内在质量。依据《浙江省医疗机构应用传统工艺配制中药制剂备案管理实施细则》[1](以下简称《细则》)的要求,本研究使用薄层色谱法(thin layer chromatography,TLC)对炙甘草、叶下珠、赤芍进行定性鉴别,高效液相色谱(high performance liquid chromatography,HPLC)对赤芍中芍药苷进行定量检测,确保临床用药安全有效。
1 仪器与试剂
1.1仪器 Thermo Fisher U3000 高效液相色谱仪(DAD 二极管阵列检测器);KQ-500DB 数控超声波清洗器(昆山市超声仪器有限公司);AL204 电子分析天平[梅特勒-托利多仪器(上海)有限公司];硅胶G 薄层板(青岛海洋化工厂)。
1.2试药 芍药苷对照品(批号110736-201741)、没食子酸对照品(批号110831-201605)、甘草苷对照品(批号111610-201607)、甘草对照药材(批号120904-201620)购于中国食品药品检定研究院,芍药苷对照品(含量测定用,批号23180-57-6,98%),购于成都普瑞法科技有限公司。白花蛇舌草、党参、垂盆草、米仁、板蓝根、焦山楂、叶下珠、乌贼骨、木瓜、五味子、土茯苓、炙甘草、赤芍等中药饮片购于浙江惠松制药有限公司。解毒软肝合剂(浙江省中西医结合医院院内制剂,批号分别为181029、181030、181031);乙酸乙酯、甲苯、三氯甲烷、甲醇、无水乙醇、甲酸、三氯化铁、乙酸、正丁醇、香草醛、硫酸、磷酸均为分析纯;甲醇为色谱纯,水为纯化水。
2 方法与结果
2.1TLC 鉴别
2.1.1炙甘草 取解毒软肝合剂20mL 蒸干,剩余残渣部分加入甲醇30mL,加热回流提取1h,之后过滤并蒸干滤液,取纯化水40mL 溶解残渣,加入正丁醇20mL 进行萃取并重复3 次,合并3 次正丁醇溶液,加入少量纯化水洗涤3 次,分离正丁醇部分,之后蒸干,剩余残渣用甲醇5mL 溶解,为供试品溶液备用[2-3]。取适量甘草苷对照品,以甲醇为溶剂,配得2mg/mL 溶液作为对照。取甘草饮片1g,同法得药材对照溶液。取解毒软肝合剂中其余饮片(除炙甘草外),根据原始处方工艺,水煎得阴性成品,并按上述方法得对照溶液,作为阴性样品。依据薄层色谱法《中国药典》2015 版四部(通则0502)[2],使用毛细管取上述四种样品溶液1~2μL,点于一块硅胶板上,展开剂为乙酸乙酯-甲酸-冰醋酸-水(15∶1∶1∶2)溶液,显色剂为10%硫酸乙醇溶液,105℃加热显色,于日光及紫外光灯(365nm)下检视。TLC 结果显示,对照药材和对照品色谱出现相似斑点,而阴性样品则未出现干扰,见图1。

图1 炙甘草薄层色谱图
2.1.2叶下珠 取解毒软肝合剂20mL 蒸干,在残渣部分加入乙醇10mL,振荡5min 后过滤,蒸干乙醇,使用1mL 乙醇溶解残渣,作为供试品备用。取适量没食子酸对照品,以甲醇为溶剂,得1mg/mL 溶液为对照[4]。取解毒软肝合剂中其余饮片(不含叶下珠),根据原始处方及工艺,水煎得阴性成品,并按上述方法得对照溶液,作为阴性样品。根据薄层色谱法《中国药典》2015 年版四部(通则0502),使用毛细管吸取对照品、供试品溶液各5μL 点于同一硅胶板上,展开剂为甲苯-乙酸乙酯-甲酸(6∶10∶1),显色剂为1%三氯化铁乙醇溶液,于日光下检视[4]。TLC 结果显示,在同一位置上,供试品与对照品显示近似斑点,且阴性样品未产生干扰,见图2。
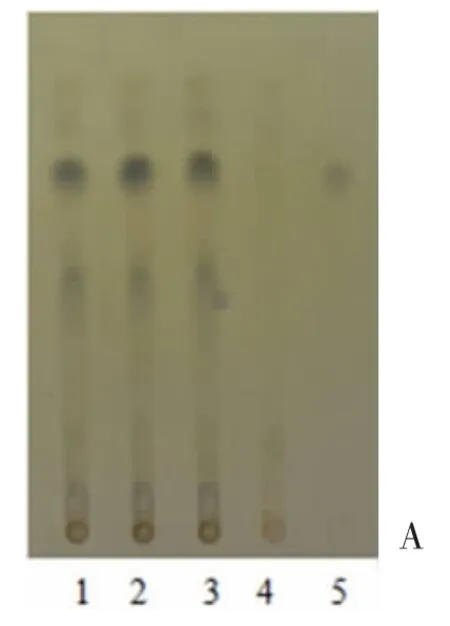
图2 叶下珠薄层色谱图
2.1.3赤芍 取解毒软肝合剂20mL 蒸干,于残渣处加入乙醇10mL,振荡5min,过滤蒸干,加入乙醇1mL 溶解残渣,作供试品备用。取适量芍药苷对照品,以乙醇为溶剂,得1mg/mL 溶液为对照。取解毒软肝合剂中其余饮片(不含赤芍),根据原始处方工艺,水煎得阴性成品,并按上述方法得对照溶液,作为阴性样品。根据薄层色谱法《中国药典》2015 年版四部(通则0502)试验,吸取对照品、供试品各2μL 于同一硅胶板上,以三氯甲烷-甲醇(5∶1)为展开剂,以5%香草醛硫酸溶液为显色剂,加热至斑点显色清晰,日光下观察[5-6]。TLC 结果显示,供试品与对照品于对应位置呈现相同颜色斑点,阴性样品则无干扰,见图3。
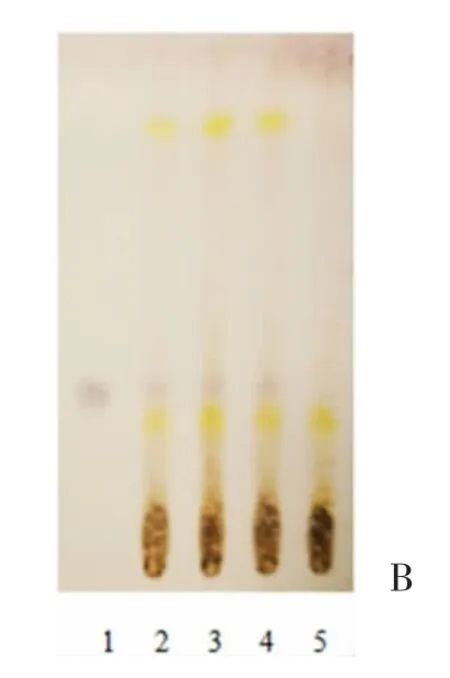
图3 赤芍薄层色谱图
2.2HPLC 含量测定
2.2.1色谱条件 色谱柱:Diamonsil C18(250×4.6mm,5μm);流动相:乙腈(A)-0.1%磷酸水溶液(B);流动相流速:1.0mL/min;梯度洗脱程序:0~30min 14% A;30~60min 14%~90%A;60~61min 90%~14% A;61~70min 14% A;进样量:10μL;检测波长:230nm;柱温:30℃。
2.2.2芍药苷对照品溶液配制 精密称取适量芍药苷对照品,以甲醇为溶剂,配成0.05707mg/mL 芍药苷对照品溶液。
2.2.3供试品制备 精密量取解毒软肝合剂1mL 于50mL 容量瓶,加入适量乙醇,超声处理(250W,40kHz)30min,放冷后定容至刻度,经0.45μm 微孔滤膜得供试品溶液。
2.2.4专属性试验 参考解毒软肝合剂处方及工艺,制得阴性样品(不含麸炒枳壳),按“2.2.3”法配制阴性溶液备用。精密吸取芍药苷对照品、供试品及阴性对照各10μL,使用HPLC 分析。结果显示,阴性样品无干扰,见图4。
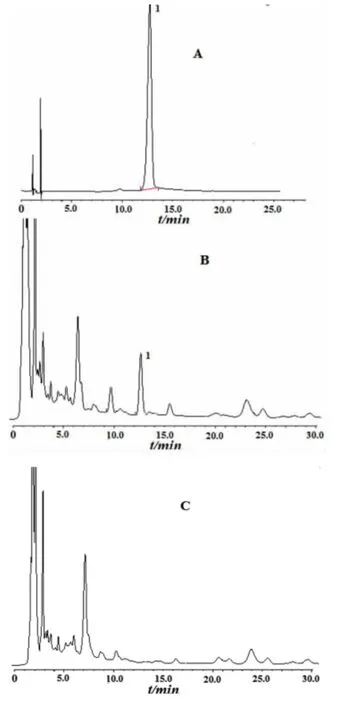
图4 高效液相色谱图
2.2.5线性关系考察 精密吸取适量“2.2.2”中芍药苷对照品溶液,逐级稀释,配成5 份对照溶液,浓度依次为:0.1177、0.2354、0.3531、0.4708、0.5885mg/mL,精密吸取上述浓度系列对照溶液10μL,使用“2.2.1”条件进行HPLC 分析。对峰面积(Y)和浓度(X,mg/mL)关系进行拟合,得线性方程:Y=1209.474X+10.589,R2=1.000,在0.1177~0.5885μg 范围内呈现良好线性关系。
2.2.6仪器精密度试验 精密吸取“2.2.2”中芍药苷对照溶液,重复进样5 次,得芍药苷峰面积RSD 为0.66%,表明本研究所用高效液相色谱仪精密度良好。
2.2.7重复性考察 取解毒软肝合剂(批号181029),按“2.2.3”法平行制备6 份待测溶液,使用HPLC 分析,得芍药苷峰面积RSD 为1.63%,表明本研究建立的含量测定方法重复性良好。
2.2.8稳定性考察 取解毒软肝合剂(批号181029),按“2.2.3”法配制供试溶液,分别于0、2、4、8、12、24h取样检测,得芍药苷峰面积RSD 为0.53%,表明解毒软肝合剂在24h 内保持稳定。
2.2.9加样回收率考察 精密量取0.5mL 解毒软肝合剂(已知含量)6 份,加入定量芍药苷标准品,根据“2.2.3”法配制待测溶液,使用HPLC 分析。计算得样品回收率平均值为99.06%,RSD=1.36%,计算公式如下,结果见表1。
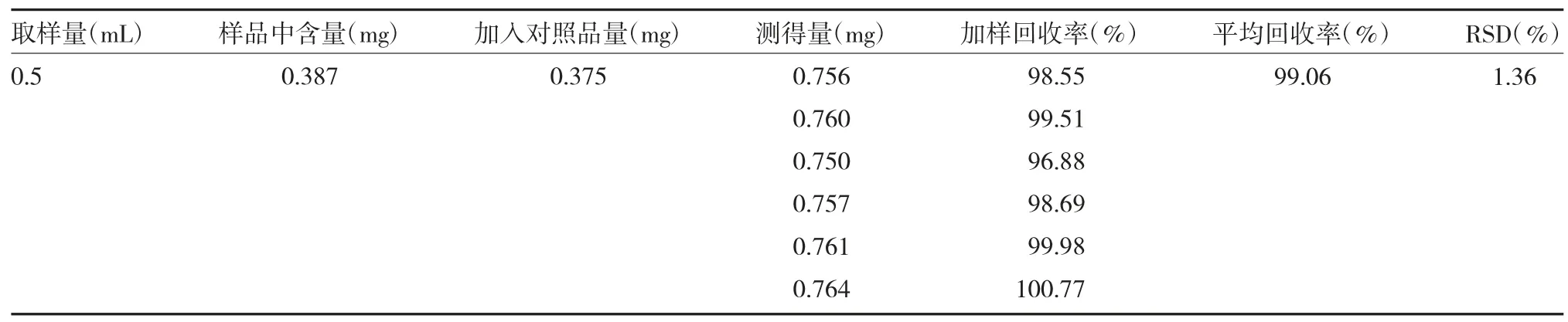
表1 芍药苷加样回收率(n=6)

2.2.10样品含量测定 取3 批解毒软肝合剂各3份,以“2.2.3”法制备待测溶液,采用HPLC 分析,以外标法计算样品含量,结果见表2。
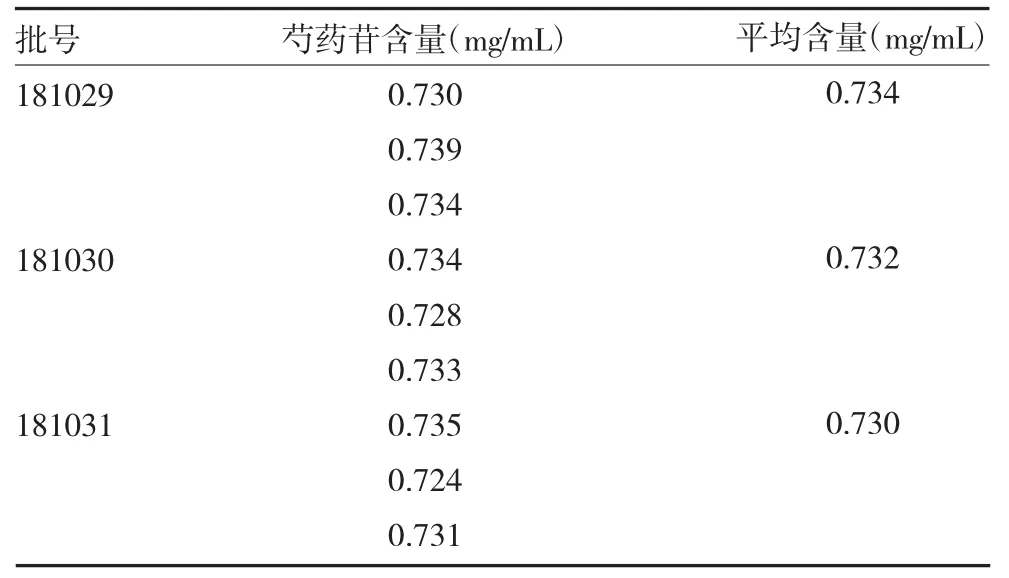
表2 样品测定结果(n=3)
3 讨论
3.1TLC 鉴别药味的选择 本研究采用TLC 对解毒软肝合剂中的炙甘草、叶下珠、赤芍、板蓝根及党参5 个药味进行研究,发现党参样品存在显著的阴性干扰,板蓝根样品的斑点不清晰,而炙甘草、叶下珠、赤芍的鉴别效果好,专属性强,分离度高,且阴性样品不存在干扰,因此选择TLC 定性鉴别炙甘草、叶下珠、赤芍,作为解毒软肝合剂的质量控制指标。
3.2TLC 鉴别湿度条件考察 湿度易影响TLC 结果,本研究比较不同湿度条件下炙甘草、叶下珠、赤芍的薄层展开效果。结果发现,在不同湿度条件下,样品斑点均能达到较好分离,方法的耐用性较好。
3.3HPLC 定量指标的选择[7]前期研究发现,君药白花蛇舌草和叶下珠在《浙江省中药炮制规范》2015版中均没有含量测定项。而预实验发现,赤芍中的芍药苷含量较高,故本品以芍药苷为指标成分,使用HPLC 法测定含量。结果表明,本研究建立的液相方法定量准确,误差小,可用于解毒软肝合剂中芍药苷含量的测定。
3.4洗脱系统的选择[8-12]解毒软肝合剂组分多,化学成分复杂,因此需要复杂的流动相体系以分离待测组分。本实验考察不同流动相系统(乙腈-水、乙腈-0.5%冰醋酸溶液以及乙腈-0.1%磷酸溶液),结果以乙腈-0.1%磷酸水溶液系统梯度洗脱分离效果最佳。液相图谱上各个色谱峰分离度较好,基线较平稳,保留时间适中。经方法学考察,线性、重复性、回收率等均符合要求。
综上所述,本研究针对解毒软肝合剂建立TLC鉴别及HPLC 含量测定相结合的质量控制体系,该体系操作简便,快捷准确,具有良好的重复性、稳定性,能够较为全面地测定解毒软肝合剂的有效成分,控制合剂整体质量,进而保证临床应用的安全有效。
