新型天冬氨酸激酶突变株构建及酶学性质表征
2022-02-16樊占青刘晓婷王哲人王亚南闵伟红
樊占青,刘晓婷,魏 贞,王哲人,王亚南,高 欣,闵伟红
(吉林农业大学食品科学与工程学院,小麦和玉米深加工国家工程实验室,吉林 长春 130118)
甲硫氨酸(Met)是一种不能在体内合成而必须由外界获取的人体必需氨基酸[1],参与生物体内的新陈代谢和蛋白质合成,是多种代谢物质的合成前体[2]。目前微生物发酵法是最有前景生产Met的方法[3],但是由于生物体内代谢路径多样复杂,各种代谢物质彼此调控,所以该方法尚未完全实现工业化生产[4]。在生物体内,天冬氨酸经过一系列代谢合成Met,而天冬氨酸激酶(aspartate kinase,AK)是该代谢途径中的第一关键限速酶[5],其活性同时受赖氨酸(Lys)和苏氨酸(Thr)的协同反馈抑制[6],导致碳流竞争激烈,Met合成供应减少,使得最终代谢产物Met不能大量积累[7-8]。目前研究发现,不同生物体内AK具有不同的存在形式,拟南芥(Arabidopsis)AK具有5 种形式,包括3种单功能AKs(AKI、AKII和AKIII)和2 种双功能AK-高丝氨酸脱氢酶(homoserine dehydrogenase,HSDH)[9-10];大肠杆菌中含有1 种单功能AKIII和2 种双功能AK-HSDHI、AK-HSDHII[11-12],此外,在一些生物体中也存在着更简单的抑制机制,詹氏酵母和嗜热菌中只含有一种形式的AK[13]。本课题组首次发现北京棒杆菌天冬氨酸激酶(Corynebacterium pekinenseaspartate kinase,CpAK)是一种新型单体别构酶,且仍受下游产物Lys和Thr的协同反馈抑制[5]。
由于CpAK在代谢途径中被下游产物协同反馈抑制,导致下游产物天冬氨酸族氨基酸产量不能大量积累,因此通过削弱或解除反馈抑制提高最终代谢产物产量成为研究热点。目前,基因敲除[14]、基因过表达[15-17]、定点突变[18-23]等基因工程技术是改造AK的主要技术手段,其中,定点突变技术的应用比较成熟广泛。Han Caijing等[5]利用定点突变技术削弱了下游产物Lys对CpAK的反馈抑制,获得酶活力提高11.32 倍的突变体A380I,为Met产量积累提供了理论依据。也有学者对抑制剂Lys和Thr周围结合位点和底物结合位点的周围关键残基进行突变研究,得到了AK活力显著提高且反馈抑制作用明显减弱的突变株R169P、R169Y、T379L和M372I/T379W,为强化AK代谢途径中下游产物碳流量的积累、构建高产Met工程菌株提供了理论依据和技术支持[16-17,24]。
目前文献报道对AK的突变改造主要围绕Lys和Thr抑制剂周围关键位点[20-26],而对催化活性中心ATP以及底物结合位点的研究相对较少。AK的一般抑制机制已从先前研究中得出结论:效应子结合诱导从底物-ATP结合的松弛状态(R状态,紧密构象)过渡到无效的紧张状态(T状态,开放构象),因此底物和ATP结合位点的构象变化均会阻碍催化作用[27]。Li Changcheng等[28]对铜绿假单胞菌AK催化域中ATP结合位点D182和R184进行双突变改造,获得酶活力降低60%的双突变株D182/R184A,证明ATP结合口袋的改变对AK活力的重要性。本研究选择AK催化中心ATP周围5 Å范围内关键残基丝氨酸(Ser227)位点,进行四突变株的构建,筛选获得高酶活力、性状良好的突变株,并结合分子动力学模拟,对其机制进行分析,旨在为优化AK代谢途径提供参考。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株、质粒与培养基
北京棒杆菌E31(吉林农业大学发酵工程实验室保藏)提供本实验AK基因;重组大肠杆菌突变株(pET-28a-T379N/A380C/G171I-AK)由吉林农业大学发酵工程实验室提供;表达宿主大肠杆菌BL21(DE3)购自北京鼎国昌盛生物技术有限责任公司。
本实验选用的培养基均为LB培养基。液体LB培养基:0.5%酵母浸粉、1% NaCl、1%蛋白胨。固体培养基则在液体培养基中加入2%的琼脂粉,溶于蒸馏水中,适宜浓度的NaOH溶液将其pH值调至7.0,121 ℃高压蒸汽灭菌20 min。
1.1.2 试剂
十二烷基硫酸钠-聚丙烯酰氨凝胶电泳(sodium dodecyl sulfate-polyacrylamide gelelectrophoresis,SDSPAGE)凝胶试剂盒、卡那霉素(kana)、丙基硫代-β-D-半乳糖苷(isopropyl-β-D-thiogalactopyranoside,IPTG)北京鼎国昌盛生物技术有限公司;DpnI消化酶、LaTaq酶TaKaRa(大连)公司;质粒DNA抽提试剂盒 生工生物工程(上海)股份有限公司;非变性镍柱及柱料美国GE公司;本实验所用引物由吉林省库美生物科技有限公司合成,基因测序由生工生物工程(上海)股份有限公司完成。
1.2 仪器与设备
梯度聚合酶链式反应(polymerase chain reaction,PCR)仪 德国Eppendorf AG公司;DYY-6C核酸电泳仪北京六一仪器厂;琼脂糖凝胶成像仪 德国耶拿分析仪器股份公司;Z36HK台式低温高速冷冻离心机 德国Hermle公司;SE260蛋白电泳仪 美国GE公司;SPECTRA Max酶标仪 美国Molecular Devices公司;WFH-201BJ蛋白印迹半干转膜仪 美国Bio-Rad公司;Scientz超声细胞粉碎仪 宁波新芝生物公司。
1.3 方法
1.3.1 定点饱和突变
首先通过Primer 5.0软件进行突变位点的引物设计,上游引物:5’-CTGGAACTTGCTGCTGTTGGCNNNAAG ATTTTGG-3’;下游引物:5’-GCGCAGCACCAAAATCT TNNNGCCAACAGC-3’。由吉林省库美生物科技有限公司合成。
根据质粒DNA抽提试剂盒说明书提取T379N/A380C/G171I-AK的质粒,以其为模板进行梯度PCR扩增全质粒基因,反应条件为:94 ℃预变性1 min;94 ℃变性1 min,56 ℃退火1 min,72 ℃延伸10 min(18个循环);72 ℃延伸10 min;4 ℃保存。
1.3.2 转宿主大肠杆菌BL21
1.3.1节完成PCR扩增,将产物用1%琼脂糖核酸电泳验证扩增成功。PCR突变产物用DpnI酶消化,反应体系为20 μL,依次加入10×T Buffer 2 μL、PCR产物2 μL、DpnI酶0.3 μL,最后用ddH2O补足,金属浴37 ℃恒温2 h。
消化结束,将2~4 μL产物转入120 μL大肠杆菌BL21感受态中,冰浴30 min,42 ℃金属浴热激90 s,继续冰浴5 min后加入900 μL不含kana的LB液体培养基,37 ℃、170 r/min摇床扩大培养2 h,5 000 r/min离心5 min后弃掉800 μL上清液,用剩余上清重悬菌体,将其均匀涂布于LB固体培养基(含kana),37 ℃倒置培养12~16 h。
1.3.3 高通量筛选及测序验证
挑取成功转入的单菌落于含kana的LB液体培养基中,180 r/min、37 ℃扩大培养12 h,加入1 mmol/L IPTG,适宜温度、130 r/min诱导表达8 h后进行高通量筛选,方法见参考文献[5],挑取酶活力显著提高的突变株加入10 mL试管培养基(含kana)于摇床180 r/min、37 ℃过夜扩大培养。将扩大培养后的突变株以菌液为模板,用克隆引物(AK引物,上游引物5’-GAATTCCATATGGC CCTGGTCGTACAGAA-3’,下游引物5’-GGAATTCTTA GCGTCCGGTGCCTGCAT-3’)进行PCR扩增验证,反应条件为:94 ℃预变性1 min,94 ℃变性30 s,58 ℃退火1 min,72 ℃延伸90 s,循环30 次,4 ℃保存[20]。将PCR产物进行1%琼脂糖核酸电泳验证,挑取目的条带明亮的菌株送到生工生物工程(上海)股份有限公司,进行全质粒基因测序验证。
1.3.4 AK分离纯化及蛋白电泳验证
测序验证成功的突变株以2%接种量于试管培养基活化12 h,吸取2 mL活化的菌液加入到100 mL LB液体培养基培养1.5~2 h后,加入终浓度为1 mmol/L IPTG诱导表达8 h,于低温高速冷冻离心机8 000 r/min离心,将上清液舍弃后收集菌体。参照朱云明等[29]实验方法制备粗酶液及纯化酶样。用SDS-PAGE和Western Blot对粗酶样及纯化样进行验证,方法参考文献[30]。
1.3.5 AK活力测定及动力学分析
AK活力以单位时间内单位质量的AK催化反应速率表示,单位为U/(mg·min),酶活力表示为1 000×A540nm。以L-天冬氨酸(L-Asp)为底物,在1 mL反应体系中加入酶样纯化液,1 mL反应体系为800 mmol/L KCl、10 mmol/LL-Asp、100 mmol/L Tris-HCl、800 mmol/L NH4OH、10 mmol/Lβ-巯基乙醇、10.4 mmol/L ATP、1.6 mmol/L MgSO4[25],保持反应体系以及其他测定条件不变,改变底物浓度(0.5、1、3、5、7、9、10、12、14、16 mmol/L)反应30 min后,于540 nm波长处测其吸光度A540nm,通过Hill方程进行非线性拟合计算AK活力。
1.3.6 AK酶学性质分析
1.3.6.1 最适温度
反应条件及反应体系不变,改变反应温度(15、20、25、26、28、30、35、40、45、50 ℃),反应30 min后终止反应,测定酶活力,每个温度检测3 组平行,最高酶活力定义为100%。
1.3.6.2 最适pH值
反应条件及反应体系不变,改变反应体系中Tris-HCl缓冲液pH值(6.0、6.5、7.0、7.5、8.0、8.5、9.0、9.5、10.0),反应30 min后终止反应,测定酶活力,每个pH值检测3 组平行,最高酶活力定义为100%。
1.3.6.3 稳定性
反应体系不变,选择最适温度和最适pH值,将酶样纯化液加入体系中,130 r/min摇床反应,间隔1 h取样一次测酶活力,每组样品检测3 组平行,将0 h酶活力定义为100%。
1.3.6.4 底物抑制剂对AK活力的影响
反应体系不变,分别加入不同浓度(终浓度分别为0.2、1.0、5.0、10.0 mmol/L)的底物抑制剂,分别为Lys、Thr、Met、Lys+Thr、Lys+Met、Thr+Met、Lys+Thr+Met,测定抑制剂对AK活力的影响,对照组加入等量蒸馏水每组样品检测3 组平行,将对照组相对酶活力定义为100%。
1.3.7 分子动力学模拟分析
以3aaw和2hmf为结构模板,同源模建获得AK三维结构,建模后丢失的原子通过VMD方法补充,同时通过局部的结构检查确定His的质子态;借助VMD的solvate plugin程序添加水溶液环境,并添加Na+或Cl-平衡体系,力场参数使用charmm36。将筛选出的高酶活力突变株T379N/A380C/G171I/S227D进行不少于110 ns的NPT自由模拟(分子动力学),分析其酶活力提高机制。
在前期实验室构建的PDB结构文件基础上,通过Discovery Studio软件分析Ser227位点突变前后与ATP及周围残基的作用力及残基间距离的变化。
1.4 数据分析
2 结果与分析
2.1 ATP结合位点的筛选
通过Discovery Studio软件对AK模型分析,Ser227位点是ATP附近5 Å范围内的关键残基;并且利用Clustal X软件进行同源序列比对,Ser227位点是保守位点,如图1所示。Han Caijing等[5]对CpAK突变体A380I进行100 ns分子动力学模拟,表明突变使A380I提高了ATP结合口袋处残基Ieu220~Glu270的均方根偏差(root mean square deviation,RMSD)值,与ATP交互作用变强,同时突变使ATP(N5)- Ser227(OG)中氢键占用率从31.74%增加到39.14%,ATP和CpAK之间氢键的占用比增多,增加了ATP和CpAK之间的结合,从而增强了CpAK的催化活性。因此本研究选择Ser227位点,设想通过饱和突变使其与ATP形成氢键,增强ATP与CpAK的结合,达到提高AK活力的预期效果。
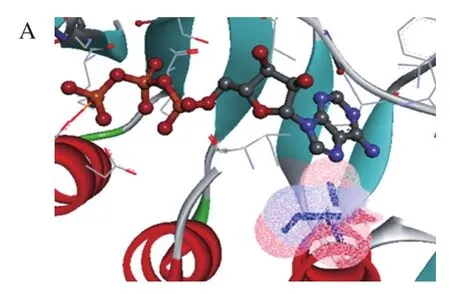

图1 ATP结合位点的筛选Fig. 1 Screening of ATP binding sites
2.2 突变株构建
以T379N/A380C/G171I(含有质粒pET-28a-AK)为模板,在Ser227引物作用下PCR扩增进行定点饱和突变。PCR产物经1%核酸电泳验证结果如图2A所示,在7 000 bp处有明亮条带,初步证明成功突变。取定量PCR产物经DpnI酶消化后转入大肠杆菌感受态细胞(BL21)中,经过高通量筛选后获得高酶活力突变株。将突变株活化后进行菌液PCR验证,如图2B所示,在1 000~2 000 bp处有明亮单一条带,与目的基因CpAK长度1 266 bp相符,进一步证明目的基因成功大量复制。将筛选的高酶活力突变株送去测序,结果如图2C所示,Ser227位点由残基Ser突变为残基Asp,最终证明突变株T379N/A380C/G171I/S227D构建成功。
2.3 SDS-PAGE和Western Blot验证
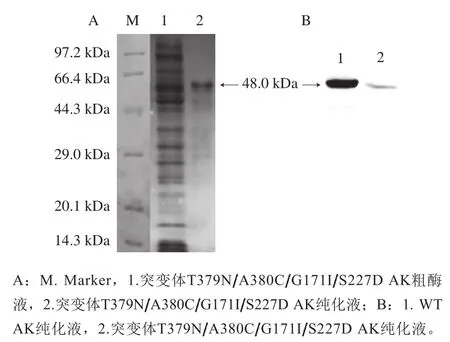
图3 SDS-PAGE(A)及Western Blot(B)结果Fig. 3 Results of SDS-PAGE (A) and Western Blot (B)
WT菌株和突变株经IPTG诱导表达目的蛋白,分别进行非变性镍柱纯化,将得到的粗酶样和纯化样进行SDS-PAGE和Western Blot验证,结果如图3所示。在48 kDa处有明显条带,证明AK蛋白表达成功。Western Blot验证结果表明,AK蛋白与抗体(His-tag27E8 Mouse mAb)特异性结合,纯化样是单一AK蛋白。
2.4 酶动力学分析
以不同浓度L-Asp为底物,对WT和突变株进行酶动力学分析,测定其酶活力。测定结果用Origin 9.0软件进行非线性拟合,根据Hill方程V=VmaxSn/(Kn+Sn)得到WT和突变株的最大催化速率Vmax。动力学结果见图4、表1,该突变株T379N/A380C/G171I/S227D的Km值减小为1.35 mmol/L,底物亲和力增大,利于AK与底物的结合,从而提高其催化效率;Vmax值为242.05 U/(mg·min),较T379N/A380C/G171I(187.88 U/(mg·min))提高到1.28 倍,较WT AK( 3.01 U/(mg·min))提高到80.41 倍,表明突变后AK的催化活力显著提高。
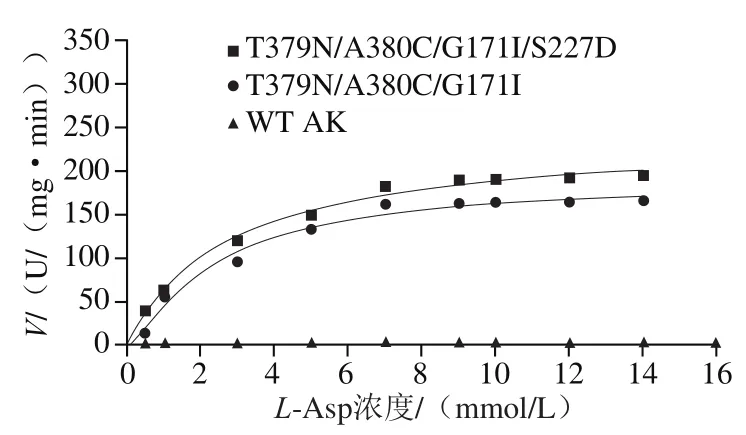
图4 WT和突变株的动力学分析Fig. 4 Kinetic analysis of WT and mutant strains

表1 WT AK和突变株的动力学参数Table 1 inetic parameters of WT AK and mutant strains
2.5 酶学性质表征
2.5.1 最适温度

图5 WT和突变株的酶学性质表征Fig. 5 Characterization of enzymatic properties of WT and mutant strains
由图5A可知,WT AK最适催化温度25 ℃,T379N/A380C/G171I AK最适催化温度28 ℃,该突变株T379N/A380C/G171I/S227D最适催化温度30 ℃,较WT和T379N/A380C/G171I分别提高5 ℃和2 ℃,表明突变增强了AK的耐热性,有效提高了AK的最适温度。当温度高于30 ℃后,该突变株的酶活力仍然较高于WT和T379N/A380C/G171I,说明多次突变有利于AK的耐高温性能的优化。
2.5.2 最适pH值
由图5B可知,WT AK最适pH值为8.0,T379N/A380C/G171I AK最适pH值为9.0,该突变株T379N/A380C/G171I/S227D最适pH值为8.5。与T379N/A380C/G171I相比,该突变株最适pH值降低0.5,耐酸性提高,同时,pH值耐受范围变大,这可能是Ser227位点由中性氨基酸Ser突变为酸性氨基酸Asp的缘故。在pH值为10时,该突变株的酶活力仍然保持在32%,较高于WT和T379N/A380C/G171I。
2.5.3 AK稳定性
相对酶活力下降到最初活力50%所用时间为酶的半衰期。选择最适温度和最适pH值,分别研究WT和突变株的稳定性。如图5C所示,WT AK的半衰期为4.9 h,而T379N/A380C/G171I AK的半衰期为3.1 h,该突变株T379N/A380C/G171I/S227D的半衰期为3.9 h。与WT AK相比,该突变体AK在反应初(0~3 h)和反应末(7~10 h)相对酶活力显著高于WT AK。与T379N/A380C/G171I AK相比,该突变株AK半衰期延长0.8 h,且相对酶活力几乎始终高于T379N/A380C/G171I AK。
2.5.4 底物抑制剂对AK活力的影响
由表2可知,WT AK受Thr和Lys两种抑制剂的协同反馈抑制,而Met对其抑制作用相对较弱。随着Thr和Lys浓度的增加,抑制作用明显增强,当Lys浓度增加到10 mmol/L时,对WT AK的抑制率大约达到39%。当两种抑制剂组合浓度为10 mmol/L时,WT AK抑制率最高为45.04%。当3种抑制剂组合浓度为10 mmol/L时,WT AK 抑制率仅比Lys+Thr组合时高4.15%,再次说明AK受Met抑制作用相对较弱,而主要受Thr和Lys的抑制,这与先前Yoshida等[31]研究一致。突变后,AK的被抑制作显著减弱,甚至被明显激活。在10 mmol/L Thr+Met、Lys+Met和Thr+Lys+Met不同组合的情况下,AK的催化活性被显著激活,T379N/A380C/G171I AK的相对酶活力分别为98.81%、100.14%和108.46%,而该突变体相对活力提高更为显著,分别为109.37%、121.25%和143.35%,且呈现明显的浓度计量依赖,这与2.4节动力学分析结果(Km减小,Vmax增大)相对应,进一步证明本研究位点Ser227是影响AK活力的关键残基位点,为后期工程菌的构建提供理论依据。
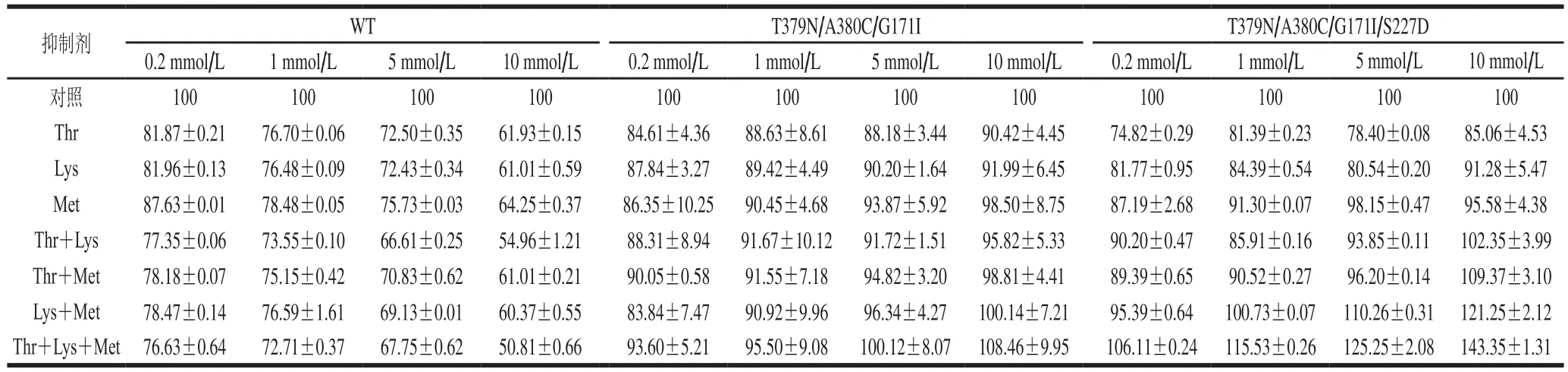
表2 不同底物抑制剂条件下WT和突变株的相对酶活力Table 2 Relative enzyme activity of WT and mutant strains under different substrate inhibitor conditions%
2.6 突变体的空间结构分析
以构建的AK模型为基础,通过Discovery Studio软件对Ser227位点突变前后与ATP和其周围残基的作用力以及距离变化进行分析。如图6A、B所示,突变前Ser227残基与ATP及周围氨基没有直接作用,突变后,S227D残基侧链向ATP延伸,与ATP和I229残基产生氢键,氢键占有率增加,连接更加紧密,更容易促进催化反应发生,这与Han Caijing等[5]研究报道一致;如图6C、D所示,突变前Ser227残基与ATP距离为2.953 Å,突变后距离缩短为2.562 Å。突变后,距离缩短,作用力增强,从而提高酶活力。
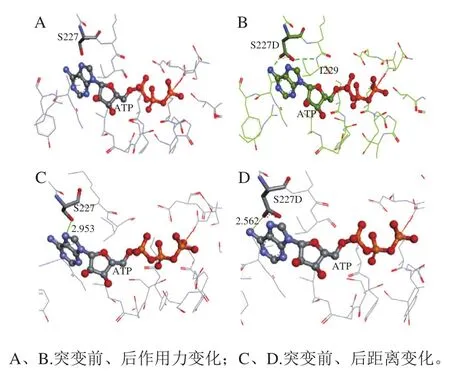
图6 Ser227位点突变前后成键变化Fig. 6 Bonding changes before and after mutation at Ser227
2.7 分子动力学模拟
对该突变体T379N/A380C/G171I/S227D AK+Asp+ATP+Lys+Thr复合物进行分子动力学模拟研究,揭示其活性提高机制。如图7A所示,复合物主链C骨架RMSD平均值为0.90 nm,体系在19 ns处达到平衡界点,之后一直平衡稳定于0.9 nm左右,说明体系在整个模拟过程中始终保持稳定状态[32]。Rg是在模拟过程中根据整个蛋白体积和性状计算出的蛋白回旋半径,反映了体系在模拟过程中的柔韧性,如图7B所示,复合物在整个模拟过程中基本平稳,平均Rg收敛于26.66 Å,整个体系柔韧性良好,这可能是由于突变使得蛋白结构发生重排趋于稳定状态。蛋白表面溶剂可及表面积(solvent accessible surface area,SASA)是描述蛋白质亲疏水性的重要参数[33],对蛋白的空间构象及其稳定性具有重要意义,由图7C可知,复合物的模拟轨迹没有较大波动,一直保持平稳,整个模拟过程平均SASA值为222.56 nm2,表明突变使得T379N/A380C/G171I/S227D 的亲水性增强,蛋白表面溶剂的亲和能力增强,从而影响其催化活性,这可能是由于突变后Asp残基侧链像ATP延伸,同时形成氢键。以上3个参数都是在蛋白质整体上进行分析研究,而均方根波动(root mean square fluction,RMSF)是从整个模拟催化过程单个氨基酸残基的相对波动情况进行分析[34]。由图7D可知,整个模拟过程中,复合物体系都表现出很好的灵活性,利于催化反应。
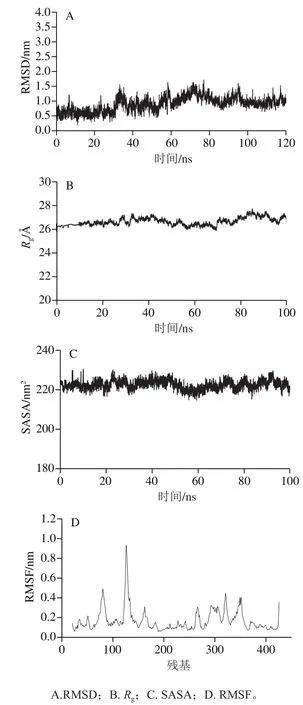
图7 T379N/A380C/G171I/S227D AK复合物分子动力学模拟分析Fig. 7 MDS analysis of T379N/A380C/G171I/S227D AK complex
3 讨 论
大量研究报道对AK的抑制剂结合位点进行突变,可以有效减弱或解除抑制作用,显著提高酶活力。Gao Yunna等[24]对抑制剂Lys周围结合位点M372和T379关键残基进行定点突变,获得酶活力提高16.51 倍的双突变体M372I/T379W AK,并通过分子动力学模拟对其机制进行分析。Min Weihong等[19]等通过虚拟筛选获得一个底物结合和催化反应的关键残基位点R169,通过定点突变技术,改造其空间结构,得到了酶活力提高4.71 倍的突变株R169Y。表明底物结合和催化反应位点的改造对AK活力提高有重要影响。魏贞等[35]研究通过对ATP周围结合位点Y198和D201进行定点饱和突变,获得性能优良、酶活力提高18.26 倍的双突变株Y198N/D201M AK。因此本研究在前期研究的基础上,继续对ATP结合位点进行突变改造,获得酶活力显著提高的突变体,进一步表明ATP结合位点的重要性。
本研究推测T379N/A380C/G171I/S227D酶活力提高的原因是T379N/A380C/G171I突变株基础上继续突变,引起ATP结合口袋重排,同时Ser227位点与ATP由非键作用变为直接氢键作用力,致使AK与ATP和底物结合更紧密,反应体系更加稳定,导致在催化过程中竞争性抑制剂不足以与底物ASP以及ATP竞争,从而减弱或解除抑制作用,显著提高AK催化活力,使得碳流竞争减弱,更多的流向下游代谢途径,进而达到高产下游代谢产物的目的,为后期构建高产蛋氨酸工程菌提供理论支持。
4 结 论
本研究对CpAK中催化域ATP周围关键残基Ser227位点进行定点饱和突变,以达到减弱或解除抑制,酶活力显著提高并获得优良多突变株的目的。通过软件Primer Premier 5 进行突变引物设计,以T379N/A380C/G171I AK为模板,经1%核酸电泳验证,成功构建突变株T379N/A380C/G171I/S227D。并将其转入宿主大肠杆菌BL21表达,经SDS-PAGE和Western Blot验证AK成功表达。动力学分析结果表明,该突变株的Vmax较T379N/A380C/G171I提高到1.28 倍,较WT AK提高到80.41 倍,酶活力显著提高,并且与底物的亲和力也明显增大,有利于与底物的结合。酶学性质研究表明:该突变株T379N/A380C/G171I/S227D的最适反应温度增至30 ℃,较WT和T379N/A380C/G171I分别提高5 ℃和2 ℃;最适pH值为8.5,较T379N/A380C/G171I(pH 9.0)减小0.5;半衰期为3.9 h,较T379N/A380C/G171I延长0.8 h;且在不同浓度、不同组合抑制剂存在时,该突变株表现出明显的激活作用,抑制效果减弱显著,相对酶活力最高达143.35%,这与动力学结果相符。同时分子动力学模拟从个体和单个氨基酸残基的变化,其中包括氢键的形成以及复合物系统中的分子间作用力,对酶活力提高进一步说明,为构建高产工程菌提供参考。
