内质网应激在抗肿瘤治疗中的作用及研究进展
2021-12-16王安琪孙国平
王安琪,孙国平
(安徽医科大学第一附属医院肿瘤科,安徽 合肥 230022)
内质网是蛋白质合成、折叠、加工、运输及参与脂质代谢的重要场所,也是细胞内储存Ca2+的主要场所。蛋白质折叠和分泌受损导致的未折叠或错误折叠蛋白在内质网腔内积累,这种现象称为内质网应激(endoplasmic reticulum stress,ERS),并引发一系列的保护性级联信号反应,称为未折叠蛋白反应(unfolded protein response,UPR)。内质网跨膜蛋白肌醇需求酶1(inositol requiring enzyme 1,IRE1)、蛋白激酶R样内质网激酶(protein kinase RNA-like endoplasmic reticulum kinase,PERK)和活化转录因子6(activating transcription factor 6,ATF6)介导3条经典的UPR通路,在缓解ERS、维持细胞内环境稳态中发挥重要作用。通常情况下,这3个传感器都通过与葡萄糖调节蛋白78 (glucose regulated protein 78,GRP78)结合保持非激活状态,但肿瘤细胞往往处于缺血、缺氧、营养物质缺乏的肿瘤微环境中,大量的未折叠蛋白或错误折叠蛋白在内质网腔内积累,GRP78与未折叠蛋白结合,并与PERK、IRE1和ATF6解离,三条信号通路激活。ERS与肿瘤的发生发展、迁移侵袭、自噬耐药、免疫逃逸[1]等密切相关。
未折叠蛋白反应是一种适应性反应,通过减少蛋白质的来源和增加蛋白质的去路减轻内质网负荷,恢复了细胞的稳态,这促进了肿瘤细胞的生存,使肿瘤恶性进展;但当过度激活的ERS超过癌细胞所能承受的阈值时,则会激活促凋亡途径诱导癌细胞死亡。故ERS相关抗肿瘤治疗可以通过两种途径:一是通过抑制UPR介导的促生存通路,二是通过诱导持续的ERS介导的促死亡通路。
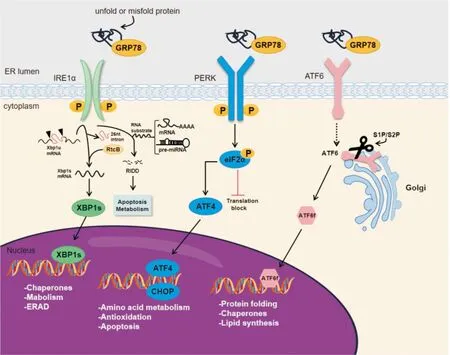
Fig 1 Unfolded protein response signaling pathway
1 抑制UPR介导的促生存通路
1.1 靶向GRP78GRP78是一种主要的内质网分子伴侣,具有Ca2+结合和抗凋亡的特性。与正常组织相比,GRP78在肝癌、胃癌、乳腺癌、肾细胞癌、子宫内膜癌及恶性胶质瘤中的表达升高,GRP78表达与肿瘤耐药、侵袭性增加和患者预后较差相关,抑制GRP78表达能明显抑制肿瘤生长,增加肿瘤细胞对药物的敏感性。例如,通过小干扰RNA(siRNA)抑制GRP78的表达增加了乳腺癌细胞的凋亡和对化疗的敏感性,也恢复了耐药乳腺癌细胞抗雌激素的敏感性[2]。GRP78最近被认为是癌症治疗的潜在靶点,体外实验中靶向GRP78的抗体在非小细胞肺癌和胶质母细胞瘤的体内外均表现出抗肿瘤活性,并能增强放疗疗效[3]。GRP78在正常成人大脑中低水平表达,但在恶性胶质瘤标本和人类恶性胶质瘤细胞系中显著升高,siRNA下调GRP78导致胶质瘤细胞生长放缓。HKH40A是WMC79的8-甲氧基类似物,是一种具有体外和体内抗肿瘤活性的合成药物,HKH40A可以通过显著降低不同来源的癌细胞株GRP78蛋白的水平从而发挥抗肿瘤活性作用。MAb159是一种抗GRP78单克隆抗体,可以特异性识别GRP78表面,触发GRP78内吞,在体内定位于肿瘤而不是正常器官,MAb159在体内外均能抑制肿瘤细胞增殖,促进肿瘤细胞死亡。研究表明MAb159可以通过抑制PI3K信号通路,代偿性激活MAPK通路,抑制肿瘤的生长和转移[4]。癌细胞中GRP78还可定位于细胞表面,细胞表面GRP78在侵袭性、转移性和耐化疗的癌症中优先过表达。GRP78在细胞表面的表达作为多种信号通路的受体,可能有很大的治疗价值,让治疗性抗体治疗成为可能。PAT-SM6就是一个例子,PAT-SM6是一种从胃癌患者中分离出来的自身抗体,该抗体可以针对GRP78的一种特殊亚型,在临床前研究中发现PAT-SM6可以抑制胃癌异种移植瘤的生长。该抗体还可特异性诱导多发性骨髓瘤细胞凋亡,而不影响正常细胞,在复发或难治性多发性骨髓瘤患者中PAT-SM6的单药治疗具有良好的耐受性和中度的临床活性[5]。在黑色素瘤中,一项药物安全性和耐受性的I期临床试验也为PAT-SM6的临床应用及进一步开发提供了有力的支持。上述研究表明靶向GRP78的表达为治疗癌症提供了新思路。
1.2 靶向IRE1α-XBP1通路作为未折叠蛋白反应中最保守的信号通路,IRE1参与调控了肿瘤发生发展的各个阶段,对IRE1通路严密调控可作为肿瘤治疗的有效手段。X盒结合蛋白1(X-box binding protein 1,XBP1)的活性形式为XBP1s,较高水平的XBP1s与更具侵袭性的乳腺肿瘤和较差的生存率相关,抑制IRE1α通过逆转ERS和重塑肿瘤微环境与抗血管生成治疗具有协同作用[6]。IRE1α表达缺失或siRNA抑制XBP1表达可抑制肿瘤增长以及减少肿瘤发生过程中的血管生成,使耐药的人胶质母细胞瘤细胞对氧化应激敏感。
IRE1α的小分子抑制剂可能具有潜在的抗肿瘤作用。MKC8866是一种小分子IRE1α RNase特异性抑制剂,通过靶向癌细胞中的IRE1通路,从而抑制了三阴性乳腺癌中MYC过表达的肿瘤生长,另外MKC8866大大增强了多西紫杉醇化疗的疗效,导致MYC过表达肿瘤的快速消退[7]。同样,在前列腺癌与横纹肌肉瘤上也观察到了MKC8866的抗肿瘤特性。Sheng等[8]研究表明MKC8866在多种小鼠临床前模型中强烈抑制前列腺癌的生长,并与现有的抗前列腺癌药物具有协同作用。在横纹肌肉瘤细胞系中观察到IRE1和PERK被激活,在加入IRE1 RNase抑制剂MKC8866或PERK抑制剂AMGEN44后,这种激活被减弱,抑制UPR可降低细胞活力、抑制细胞增殖及集落形成[9]。STF-083010是IRE1α核糖核酸内切酶抑制剂,可特异性阻断IRE1α-XBP1信号通路。与非耐药的乳腺癌细胞相比,耐药的乳腺癌细胞XBP1在mRNA和蛋白水平上上调,STF-083010处理后重新建立了耐药乳腺癌细胞对他莫西芬的敏感性,STF-083010和他莫西芬联合治疗可以显著延缓异种移植乳腺肿瘤模型中乳腺癌的进展。在卵巢癌中STF-083010也通过激活caspase -12、-3和Bax/Bcl-2蛋白的表达,降低细胞增殖,诱导细胞凋亡[10]。STF-083010在多发性骨髓瘤中也具有抗肿瘤活性。另一种IRE1α核糖核酸内切酶结构域小分子抑制剂MKC3946,通过抑制XBP1的剪接,抑制了多发性骨髓瘤细胞的生长,并且增强了硼替佐米和17-AAG诱导的细胞凋亡。不同类型的IRE1α特异性抑制剂的抗肿瘤作用还在胰腺癌、肥大细胞白血病、急性髓系白血病中进行了相关研究,这些IRE1α特异性抑制剂通过抑制增殖、触发凋亡,以剂量和时间依赖性抑制细胞生长,并且与原有的抗癌药物具有协同效应。
抑制IRE1α也可提高抗肿瘤免疫。在从卵巢癌患者标本中分离的T细胞中,XBP1的上调与T细胞向肿瘤浸润减少和IFNG mRNA表达降低有关。患有卵巢癌且T细胞XBP1选择性缺失的小鼠抗肿瘤免疫能力更强,并且延缓了癌症的恶性进展,提高了总生存率。故控制ERS或靶向IRE1α-XBP1信号通路可能有助于恢复肿瘤宿主T细胞的代谢适应性和抗肿瘤能力[11]。这些数据表明靶向阻断IRE1α-XBP1信号通路可能是一种潜在的抗癌治疗策略。
1.3 靶向PERK-eIF2α-ATF4通路PERK蛋白在肝癌组织中高表达且与肿瘤的侵袭能力和索拉菲尼抵抗密切相关[12]。PERK-真核细胞翻译起始因子2α(eukaryotic initiation factor-2α,eIF2α)-活性转录因子4(activating transcription factor4,ATF4)信号通路是导致癌症生长和耐药的原因。PERK缺乏的细胞对ERS敏感,抑制PERK可以抑制缺氧耐药癌细胞。PERK对放疗的疗效也有显著影响,溶酶体相关膜蛋白3 (lysosomal associated membrane protein 3,LAMP3)是UPR的PERK-ATF4轴诱导的,LAMP3在乳腺癌放疗患者中有预后价值。敲除PERK-ATF4-LAMP3轴或化学抑制PERK可以使MDA-MB-231乳腺癌细胞对放疗敏感,下调LAMP3还会使耐药乳腺癌细胞对它莫西芬重新敏感[13]。
PERK、eIF2α的小分子抑制剂也可能具有潜在的抗肿瘤作用。PGSK2656157是一种ATP竞争性的PERK酶活性抑制剂,可以通过改变氨基酸代谢,降低血管密度和血管灌注,抑制小鼠体内多种人肿瘤异种移植瘤的生长。GSK2606414是一种口服有效的选择性PERK抑制剂,可以抑制细胞中PERK的激活,并抑制小鼠体内的人类肿瘤异种移植瘤的生长,用其处理乳腺癌细胞提高了癌细胞对放疗的敏感性[14]。ISRIB是一种PERK-eIF2α活性抑制剂,Nguyen等[15]利用小鼠和人前列腺癌模型,发现在晚期前列腺癌中PERK-eIF2α被选择性激活,推动了肿瘤的侵袭性进展,ISRIB通过靶向蛋白的合成,可选择性地触发对侵袭性转移性前列腺癌的细胞毒性,利用ISRIB靶向eIF2α信号通路可以限制肿瘤细胞的转移和侵袭。
抑制内质网应激PERK通路,可以提高抗肿瘤免疫。骨髓来源抑制细胞(myeloid-derived suppressor cells,MDSCs)是多种疾病相关病理的中心驱动因素,这些细胞的主要功能特征是抑制免疫反应以抑制炎症。在MDSCs中,抑制PERK表达可诱导抗肿瘤T细胞,并与免疫治疗具有协同作用[16]。通过以上研究结果可知,抑制PERK相关信号通路可能是癌症治疗的一个靶点,研究其相关机制可能为肿瘤治疗提供新方向。
1.4 靶向ATF6通路ATF6在UPR中发挥细胞保护作用,这是细胞在ERS下生存所必需的。ATF6α的激活促进了癌细胞的存活。有研究发现ATF6α在宫颈癌细胞中表达较高,抑制ATF6可以降低细胞活力和迁移,通过抑制Bcl-2和增加caspase-3促进细胞凋亡[17]。ATF6还与肿瘤耐药相关,在1104例高级别浆液性卵巢癌组织中,DNA结合抑制因子1(ID1)和ATF6的表达存在显著相关性,高表达ID1或ATF6的患者对铂类治疗耐药,整体生存率和无进展生存率较差[18]。Gallagher等[19]研究鉴定了ATF6信号的选择性抑制剂,即小分子Ceapins,这是一类吡唑酰胺,它可以阻断ATF6α信号通路来响应ERS。Ceapins使细胞对ERS敏感,而不影响非应激细胞的活力,高度特异性抑制ATF6α信号,而且不抑制高尔基蛋白酶或其他UPR分支,因此有潜力用于开发以诱导癌细胞死亡。Bu等[20]研究指出,褪黑素或siRNA抑制了ATF-6表达显著增加C/EBP同源蛋白(C/EBP homology protein,CHOP)表达水平,进而抑制环氧合酶-2的表达,增强ERS条件下肝癌细胞凋亡。故靶向阻断ATF6α信号通路可能是一种潜在的抗肿瘤治疗策略。
1.5 靶向ERS相关自噬肿瘤细胞发生ERS不仅可诱导UPR,还可诱导自噬。自噬作为一种减轻ERS并允许细胞存活的方式促进了肿瘤的生长。在肝癌细胞中,ERS增强了肝癌细胞的自噬通量,有助于维持细胞活力[21]。ERS依赖性自噬还可增强肝癌细胞耐药,研究发现锌指蛋白263在肝细胞癌患者和细胞系中表达上调并且通过激活ERS相关的自噬增加了肝癌细胞的化疗耐药性[22]。在黑色素瘤细胞中,BRAFi诱导的细胞自噬可以促进癌细胞在ERS条件下的增殖和转移;在宫颈癌细胞中,TAW诱导的自噬能对抗细胞的凋亡。靶向ERS相关自噬具有潜在的抗肿瘤作用,如黄芪多糖可通过下调PI3K/AKT信号通路的自噬而在肺癌中发挥抗肿瘤作用[23]。褪黑激素通过下调细胞自噬增强了索拉非尼对人肝癌细胞系的抗肿瘤作用[24]。在三阴性乳腺癌中,紫杉醇耐药MDA-MB-231乳腺癌细胞系自噬活性增强,抑制自噬增强了耐药细胞的死亡。用自噬抑制剂氯喹处理宫颈癌HeLa细胞后促进了紫杉醇诱导的细胞凋亡。Zhou等[25]研究发现,低浓度的褪黑激素通过PERK-ATF4-Beclin1通路抑制自噬,增加了肝癌患者对索拉非尼的敏感性,其研究表明在肝细胞癌患者标本中,自噬相关蛋白Beclin1的表达与PERK的表达高度相关,联合表达PERK和Beclin1的患者病情更严重,总生存时间更短。通过特定抑制剂抑制ERS和自噬时,索拉非尼诱导细胞凋亡增加,选择性敲除PERK和ATF4表达降低了索拉非尼诱导的自噬活性,且沉默ATF4抑制了Beclin1的表达。
2 增强ERS介导的促死亡通路
持续强烈的ERS通过CHOP、c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)和caspase-12 3条信号通路介导促死亡通路是治疗肿瘤的另一策略。其中,CHOP和JNK信号通路是ERS和细胞凋亡之间相互联系的重要分子,caspase-12是caspase家族中唯一一个仅在ERS途径中被激活,而不参与线粒体凋亡途径和死亡受体途径的分子,是细胞凋亡命令的执行者。CHOP通过干扰细胞周期、损伤相关基因、抑制内质网蛋白折叠能力,而在细胞凋亡中具有重要功能。在肺癌及肝癌的小鼠模型中,观察到CHOP的特异性缺失会导致肿瘤的生长,促进肿瘤的发展,故上调CHOP表达可能是一个潜在的抗癌治疗靶点。白藜芦醇通过提高胞质Ca2+水平,激活PERK、eIF2α和上调CHOP,选择性地诱导吉非替尼耐药的非小细胞肺癌细胞死亡。褪黑激素通过下调PI3K/AKT通路、增加CHOP水平来减弱ERS诱导的人肝癌细胞对阿霉素的耐药性[26]。ERS相关途径通过JNK信号通路在肿瘤治疗中发挥重要作用。激活的IRE1α与肿瘤坏死因子受体相关因子2 (TRAF2)相互作用,导致凋亡信号调节激酶1 (ASK1)和JNK的增加,进而诱导细胞凋亡。敲除小鼠的JNK基因后导致肿瘤数量增加并且加快了肿瘤的生长速度,表明JNK通路可能通过抑制肿瘤细胞的生成或诱导肿瘤细胞凋亡,防止肿瘤的发生。IRE1α-JNK-JUN通路可恢复KRAS突变的结肠癌细胞系对MEK抑制剂的敏感性。
另外有几种ERS诱导剂有潜力用作抗癌药物治疗。Eeyarestatin I (EerI)是一种内质网应激相关降解途径(ER-associated degradation,ERAD)抑制剂,该抑制剂阻断未折叠蛋白从内质网进入细胞质降解,它与硼替佐米有类似的抗肿瘤和生物活性,并可与硼替佐米协同作用,通过诱导ERS,对癌细胞产生细胞毒性。用EerI处理人黑色素瘤细胞时,GRP78在胞质中积累,CHOP在细胞核中积累,与单药治疗相比,EerI和顺铂联合治疗导致黑色素瘤细胞死亡显著增加。衣霉素(TM)是一种天然产生的抗生素,它通过阻断内质网和高尔基体中n-链寡糖生物合成的第一步来抑制蛋白质糖基化。衣霉素在各种癌症中表现出明显的抗肿瘤活性,其通过UPR诱导ERS,从而促进细胞凋亡。TM诱导的ERS增加了乳腺癌细胞对放疗的敏感性,通过调控Akt/NF-kB信号通路抑制乳腺癌细胞生长和转移,并且抑制MCF7乳腺癌干细胞的侵袭性[27]。TM诱导的ERS,可以通过抑制N-糖基化抑制头颈部癌细胞的发生;通过激活mTORC1促进前列腺癌细胞凋亡;通过诱导E-cadherin介导的细胞粘附,使未分化结肠癌细胞的增殖受到抑制。TM还可以增强癌症细胞对药物的敏感性,如增强顺铂对肺癌生长的抑制作用,增强人非小细胞肺癌细胞对厄洛替尼的敏感性等。Brefeldin A 是一种细胞内蛋白质转运抑制剂,可阻断蛋白质向高尔基体的运输使蛋白质在内质网中积累,诱导ERS。研究表明Brefeldin A能有效诱导结肠癌、乳腺癌细胞凋亡,还能降低MMP-9活性,提示其在结肠癌、乳腺癌发生和转移阶段可有效抑制其进展[28]。毒胡萝卜素(TG)是ERS源,启动UPR介导的凋亡,对多种肿瘤细胞系具有潜在的抗癌活性。最近Linder等[29]证明,TG处理的人前列腺癌和结肠癌细胞株,通过死亡受体5、MAP1LC3B的非自噬功能以及UPR等途径诱导细胞死亡。另外在食管鳞癌、黑色素瘤及肾上腺皮质癌中,也观察到TG通过激活ERS诱导癌细胞凋亡,提高了抗癌药物治疗疗效。蛋白酶体抑制剂也可能通过不同途径加重内质网负荷,引起内质网应激介导的细胞死亡。有研究报道,硼替佐米可能通过ERS导致的活性氧(ROS)增加使细胞产生凋亡从而抑制多发性骨髓瘤[30]。因此,通过诱导持续的ERS介导的促死亡通路为ERS相关的抗肿瘤治疗提供了另一途径。
3 结论与展望
UPR是细胞应对ERS的一种重要的保护机制。正常细胞所处的内环境适宜细胞生长,不需要利用UPR,而肿瘤细胞处在肿瘤微环境中需要依赖于UPR信号以在不利环境中生存,因此,靶向UPR相关信号通路可能有利于特异性地消除癌细胞。同时,过度激活ERS超过癌细胞所能承受的阈值,以激活细胞的促凋亡通路也可导致细胞死亡。然而我们需要思考的是,UPR既具有促生存效应,也可以激活下游的促凋亡通路,单纯抑制UPR信号通路可能会抑制其激活的促凋亡通路,这样反而促进了癌细胞的存活。因此,我们希望找一种方法,既可以抑制UPR的促生存效应,又对下游的促凋亡通路不产生影响,使癌细胞走向凋亡。另外,不断增强ERS的强度对正常细胞也可能产生影响,故我们希望找到一个临界值,既促进癌细胞凋亡,又对正常细胞不产生影响,以更好地促进癌症的治疗。尽管近年来国内外关于ERS方面的研究已经取得了一些成果,但并不完善,仍然存在许多问题。因此,研究者仍需不断地阐明ERS在肿瘤治疗中的作用机制,以便为肿瘤治疗提供新的思路。
