内质网自噬及其与疾病的关系研究进展
2022-01-18张慧芳黄自能陈蔚孙林刘伏友肖力
张慧芳,黄自能,陈蔚,孙林,刘伏友,肖力
1中南大学湘雅二医院肾内科,长沙 410000
自噬是消除聚集的蛋白质及受损细胞器的过程,对于缓解应激、维持细胞稳态及细胞分化等至关重要[1-2]。既往研究认为,自噬对细胞内各种组分的降解是非选择性的,但目前研究发现,自噬可特异性吞噬多种细胞器,如线粒体、脂滴及内质网等[3-5]。内质网是细胞内最大的内膜系统,在机体的基本生命活动中发挥重要作用。一方面,内质网参与了蛋白质和脂质的生物合成、离子稳态、新生蛋白的质量控制以及细胞器之间的交流,维持细胞的基本功能;另一方面,多种因素可诱发内质网稳态失衡,启动细胞凋亡程序。内质网持续更新可适应不同的细胞需求,而自噬在这个过程中起着重要作用。在细胞应激状态下,内质网通过未折叠蛋白反应诱导分子伴侣和相关酶的表达,促进蛋白质折叠,维持内质网稳态。一方面,内质网相关降解途径可识别错误折叠蛋白,促进其转位至胞质被蛋白酶体降解;另一方面,某些无法转运至胞质的错误折叠蛋白质需要通过内质网自噬降解。内质网自噬的主要生理功能是通过溶酶体降解未折叠蛋白质、错误折叠蛋白质和应激的内质网,维持细胞稳态[6]。2005年有研究报道,在酵母中,饥饿可促进内质网通过自噬传递至液泡[7]。2015年,Khaminets等[8]发现了内质网自噬受体FAM134B后,内质网自噬相关研究有所进展。越来越多的研究表明,内质网的清除是一个特异性的过程,目前已发现多种内质网自噬受体,但其调控机制仍需进一步研究。内质网自噬与多种疾病密切相关,如神经退行性疾病、感染和肿瘤等。研究内质网自噬的分子调控机制有助于了解疾病的发病机制,寻找新的治疗策略。本文对内质网自噬的机制及其与疾病的关系展开综述。
1 内质网自噬及其机制
1.1 内质网自噬受体介导自噬体对内质网的特异性识别 内质网自噬可分为大自噬、小自噬和囊泡传递三种类型。大自噬是指内质网片段和其他内质网腔内成分被自噬体包裹,当自噬体的外膜与溶酶体融合后,自噬体的内膜及其内容物如内质网片段被溶酶体降解。小自噬指部分内质网片段直接被溶酶体或晚期内体吞噬降解[9]。囊泡传递指包含未折叠蛋白质或错误折叠蛋白质的囊泡从内质网出芽,直接与溶酶体融合[10]。目前研究以大自噬为主,本文主要介绍由自噬相关蛋白(autophagy-related gene,Atg)和内质网自噬受体介导的大自噬。
多种自噬相关蛋白在自噬的不同阶段发挥着重要作用,如自噬体的形成和成熟、自噬体对货物的识别以及自噬体与溶酶体的融合等。研究发现,Atg8在自噬体对内质网的特异性识别中发挥重要作用。自噬体膜上的Atg8与内质网自噬受体结合,促进自噬体沿着降解靶标扩展,最终实现对受损内质网碎片的包裹。目前发现,哺乳动物有6种Atg8家族蛋白:微管相关蛋白1轻链3(microtubule associated protein 1 light chain 3,MAP1LC3)亚家族LC3A(两种异构体)、LC3B和LC3C,以及GABA A型受体相关蛋白(GABA type A receptorassociated protein,GABARAP)亚家族GABARAP、GABARAPL1和GABARAPL2[11]。
哺乳动物中内质网自噬受体通过LC3相互作用区域(LC3-interacting region,LIR)或GABARAP相互作用区域(GABARAP-interacting motif,GIM)直接募集自噬体上的LC3或GABARAP。酵母中内质网自噬受体通过Atg8相互作用区域(Atg8-interacting motif,AIM)与自噬体上的Atg8结合,促进内质网与自噬体的融合。相互作用蛋白组学研究发现,有8种内质网跨膜蛋白可作为内质网自噬受体,包括FAM134B、Sec62、长链RTN3(RTN3 long isoform,RTN3L)、CCPG1、ATL3、TEX264、Atg39和Atg40(前6种存在于哺乳动物,后2种存在于酵母)(图1、表1)。最新研究发现,可溶性蛋白质CALCOCO1和Epr1也可作为内质网自噬受体(前者存在于哺乳动物,后者存在于酵母)。这些内质网自噬受体在不同的组织、细胞亚区域、生理和病理条件下发挥作用,其多样性和潜在机制有待进一步深入研究。

图1 哺乳动物和酵母中内质网自噬受体的结构Fig.1 Structure of reticulophagy receptors in mammals and yeast

表1 哺乳动物中内质网自噬受体的特点Tab.1 Characteristics of reticulophagy receptors in mammals
FAM134B是FAM134蛋白家族成员,通过羧基端的LIR结构域与LC3B或GABARAP结合,诱导片状内质网的降解。FAM134B跨膜区域的浆膜蛋白同源结构域(reticulon-homology domain,RHD)有两个楔形的跨膜螺旋夹和两个两亲性的螺旋结构,通过寡聚化诱导膜弯曲及蛋白分选[12]。内质网应激条件下,活化的钙调蛋白激酶-2(calcium/calmodulindependent protein kinase 2,CAMK2)磷酸化RHD,增强FAM134B在内质网发生碎片化时的寡聚化和活性,促进内质网自噬[13]。RHD结构域被破坏和FAM134B缺失可引起片状内质网大量扩展[8]。饥饿可通过激活CCATT增强子结合蛋白β(CCATT enhancer binding proteins,C/EBPβ)诱导FAM134B-2表达,促进FAM134B-2与自噬体及溶酶体的融合[14]。FAM134B介导的内质网自噬具有重要的生理功能。成纤维细胞生长因子18(fibroblast growth factor 18,FGF18)可通过转录因子TFEB/TFE上调FAM134B的表达,特异性激活内质网自噬,促进青鳉鱼软骨细胞蛋白分泌、软骨生长和骨钙化[15-16]。Forrester等[17]发现,FAM134B和内生内质网驻留的钙联蛋白(calnexin,CANX)参与了原骨胶原的质量控制:CANX与FAM134B相互作用,同时作为共受体识别内质网腔内错误折叠的原骨胶原,然后FAM134B与自噬体膜上的LC3结合,将包含CANX和原骨胶原的内质网传递至溶酶体降解。然而,内质网自噬过度也可引起细胞损伤和相关疾病。FAM134B过表达可导致HeLa细胞内质网应激、未折叠蛋白反应和细胞死亡[18]。
Sec62是哺乳动物转位复合物Sec61/Sec62/Sec63的成分之一,可羧基端胞质区含有一个LIR结构域,可作为内质网自噬受体参与内质网应激的恢复过程,特异地传递过量的内质网片段至自噬体,恢复内质网的稳态[19]。球形脂联素通过激活AMPK上调Sec62介导的内质网自噬,减轻慢性间断性低氧引起的内质网应激和H9C2心肌细胞凋亡[20]。在恶性疟原虫和植物中,Sec62的AIM结构域也可与Atg8相互结合,参与内质网自噬。拟南芥Sec62突变体生长受限、授粉异常、繁殖能力下降[21-22]。
浆膜蛋白RTN3L(the long isoform of RTN3,RTN3L)塑造了内质网的管状区域,其氨基端有6个AIM结构域,跨膜区域有一个RHD结构域,可作为特异性受体与LC3/GABARAP结合。RTN3L的寡聚化足以触发内质网的碎片化,协助管状内质网传递至溶酶体,且不依赖同样具有RHD结构域的FAM134B[23]。玉米胚乳中存在RTN1和RTN2的表达,可以剂量依赖的方式重塑内质网结构。内质网应激可促进RTN1和RTN2与Atg8相互作用,调节大自噬,维持内质网稳态[24]。
作为一种内质网驻留的跨膜蛋白(无其他已知的生理功能),CCPG1有一个LIR结构域和两个FIP200相互作用结构域(FIP200 interacting region,FIR),对GABARAP和LC3具有双重亲和力。Smith等[25]发现,CCPG1是内质网应激的效应子,可通过与Atg8和FIP200相互作用参与内质网自噬,维持胰腺腺泡细胞内质网蛋白稳态,防止蛋白质聚集和未折叠蛋白反应过度激活引起的细胞死亡。
Atlastins(ATL1、ATL2、ATL3)是一类膜结合、动力蛋白样GTP酶,参与了管状内质网的融合。饥饿状态下,ATL3通过氨基端的两个GABARAP互作结构域特异性结合自噬体的GABARAP,介导管状内质网的降解[26]。Liang等[27]发现,ATL2位于内质网自噬受体FAM134B的下游,其缺失可抑制FAM134B过表达引起的片状内质网自噬。目前研究发现,FAM134B主要介导片状内质网的自噬,表明ATL2也可能参与片状内质网的重塑,协助分离FAM134B标记的内质网,促进自噬体与内质网的融合。
内质网膜蛋白TEX264有一个跨膜结构域和一个LIR结构域,参与管状内质网的降解。与其他自噬受体相比,TEX264与LC3和GABARAP家族蛋白的相互作用更高效,表达更广泛。TEX264约占饥饿状态下自噬流量的50%[28]。单独敲除TEX264可导致内质网自噬显著减弱;同时敲除TEX264、FAM134B和CCPG1时,内质网自噬基本完全消失[29]。
Atg39和Atg40可作为酿酒酵母(Saccharomyces cerevisiae)特异性的内质网自噬受体和核自噬受体。Atg39定位在核周内质网和核被膜,诱导部分核的自噬隔离。Atg39的氨基端含有一个AIM和一个Atg11互作结构域,以及一个跨膜结构域。在氮剥夺的情况下,Atg39依赖的核周内质网自噬和核自噬可促进细胞存活。Atg40在皮层和胞质内质网中含量丰富,其氨基端含有一个AIM结构域和两个跨膜结构域[30]。在饥饿条件下,自噬体的Atg8与Atg40多价结合,诱导Atg40的浆膜蛋白样结构域产生高度弯曲的内质网,并且包装这些结构域进入自噬体[31]。
CALCOCO1是一种可溶性的内质网自噬受体,参与毒性蛋白质和饥饿应激引起的管状内质网降解,其羧基端含有一个FFAT[two phenylalanines(FF) in an acidic tract (AT),FFAT]样结构域和一个UIM(ubiquitin-interacting motif,UIM)结构域,氨基端含有一个LIR结构域。CALCOCO1通过FFAT样结构域与内质网膜蛋白VAPA和VAPB相互作用,通过LIR和UIM结构域与Atg8直接结合,触发大自噬。与LC3相比,CALCOCO1倾向于与GABARAP结合。敲除CALCOCO1可导致应激状态下的内质网自噬受损[32-34]。
Epr1是可溶性的内质网自噬受体,由内质网应激诱导产生。Epr1的羧基端含有一个AIM和一个FFAT结构域。Epr1通过FFAT结构域与内质网膜蛋白VAP相互作用定位在内质网上。同时,Epr1通过AIM结构域与Atg8相互作用。内质网应激时,Epr1可被未折叠蛋白反应调节因子肌醇依赖酶1α(inositol-requiring enzyme 1α,IRE1α)上调。Epr1缺失可导致细胞对内质网应激的耐受力下降,死亡率上升[35]。
1.2 Lst1/Sec24C-Sec23介导自噬体对内质网的隔离 COPII货物衔接复合物Lst1/Sec24C-Sec23除具有分泌功能外,还可靶向管状内质网特定区域进行降解[36](图2)。Lst1/Sec24C-Sec23通过与内质网跨膜蛋白(内质网自噬受体或分泌蛋白)的细胞质结构域相互作用来分选不同的货物,引导货物进入分泌途径或内质网自噬途径,这两种不同的内质网运输途径由各自的应激反应调节。Lst1/Sec24C介导的内质网自噬由营养剥夺或易聚集蛋白质介导。在酵母中,上调内质网自噬受体Atg40可诱导Lst1-Sec23与Atg40的胞质结构域相互作用,并包装内质网进入自噬体,介导自噬体对内质网的隔离。在Torin2处理的U2OS细胞中,Lst1的哺乳动物同源类似物Sec24C参与了FAM134B和RTN3L介导的内质网自噬,表明Lst1的功能可能是保守的。但哺乳动物中Sec24是否直接与FAM134B或RTN3L结合而介导自噬体对内质网的隔离仍需进一步研究。包含Lst1/Sec24C-Sec23的内质网自噬位点需要内质网跨膜蛋白Lnp1稳定管状内质网的连接,其缺失可阻碍Atg40与Atg11(参与自噬体的形成)结合以及内质网进入自噬体[37]。
1.3 泛素化参与内质网自噬 Liang等[38]通过全基因组测序发现泛素化基因参与了内质网自噬(图2)。内质网膜蛋白三重基序蛋白13(tripartite motif,TRIM13)(E3泛素连接酶)可自身泛素化并募集p62,同时,内质网驻留的分子伴侣通过精氨酰转移酶1实现精氨酰化,这些精氨酰化的分子伴侣诱导p62的寡聚化,寡聚化的p62与自噬体上的LC3相互作用,表明p62可能作为一种内质网自噬受体。
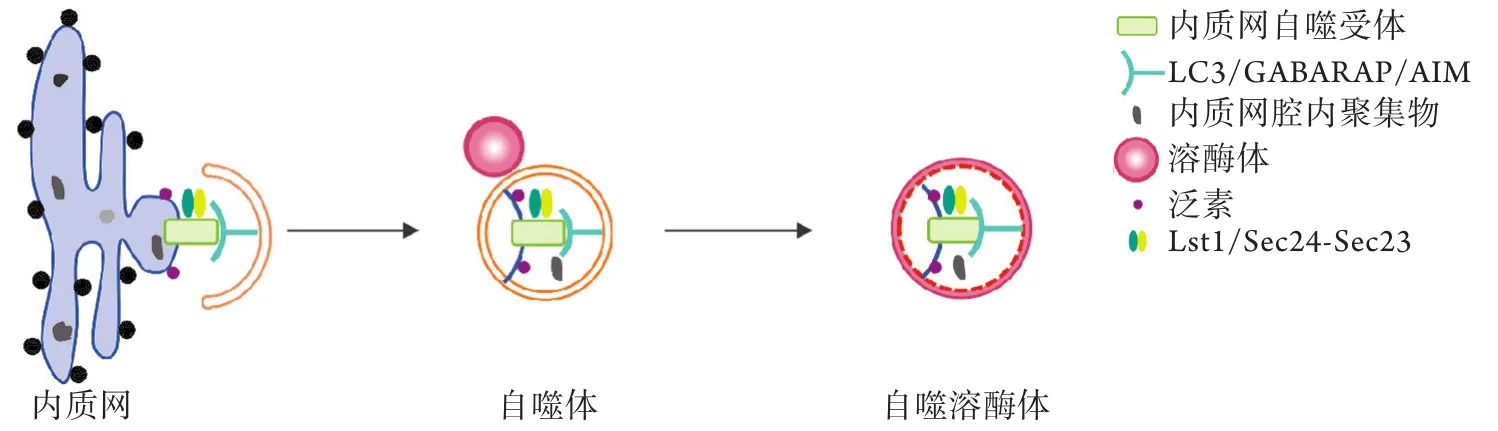
图2 哺乳动物和酵母中内质网自噬的机制Fig.2 Mechanisms of reticulophagy in mammals and yeast
类泛素化Ufm1修饰也参与了内质网自噬[38]。与泛素化修饰类似,Ufm1修饰由E1样活化酶(Uba5)、E2样结合酶(Ufc1)和E3样连接酶(UFL1)催化完成。DDRGK1是Ufm1修饰的重要调节因子,调控并维持UFL1的活性[39]。饥饿条件下,内质网膜蛋白DDRGK1将UFL1连接酶携带到内质网表面,类泛素化修饰内质网上的RPN1和RPL26(两者均为核糖体结合蛋白),促进内质网自噬,抑制IRE1α的表达和未折叠蛋白反应。此外,RPN1可与Sec61/62/63转位复合物相互作用,但Ufm1修饰是否影响内质网自噬受体Sec62的表达水平与活性,以及促进内质网自噬的具体机制有待研究。上述研究结果提示泛素化在内质网自噬中发挥了重要作用。
2 内质网自噬与疾病的关系研究进展
2.1 内质网自噬与糖尿病 最近一系列研究发现,内质网自噬参与了胰腺胰岛部和外分泌部正常功能的维持。胰岛素基因突变所致的青少年糖尿病(mutant INS-gene-induced diabetes of youth,MIDY)与胰腺胰岛部功能障碍有关。突变产生的Akita胰岛素原在内质网中形成聚合物并包被正常的胰岛素原,阻止其从内质网中分泌。Cunningham等[40]发现,上调RTN3可促进Akita胰岛素原的清除,从而保证内质网输出正常胰岛素原,具有潜在的治疗价值。胰腺外分泌部内质网腔内蛋白聚集可导致未折叠蛋白反应过度激活及组织损伤,而未折叠蛋白反应可同时诱导CCPG1介导的内质网自噬,维持胰腺内质网蛋白质稳态。CCPG1敲除会导致小鼠胰腺外分泌部的粗面内质网过度膨胀,且腔内浓缩的包含物颗粒增多,炎性细胞浸润增加,内质网应激分子BiP、Chop、Grp94和sXBP1的表达水平升高[25]。
2.2 内质网自噬与肾脏损伤 内质网自噬可介导碲化镉引起的肾脏损伤。体内研究发现,小鼠静脉注射碲化镉后,肾脏是碲化镉分布最多的器官之一,并出现功能障碍,且肾脏存在未折叠蛋白反应和FAM134B介导的内质网自噬。体外实验发现,碲化镉可破坏HEK细胞内质网的超微结构,使用抑制剂或siRNA阻断未折叠蛋白反应可缓解碲化镉触发的内质网自噬。此外,抑制未折叠蛋白反应或FAM134B依赖性的内质网自噬可恢复HEK细胞的活力[41]。
2.3 内质网自噬与神经系统疾病 内质网自噬对于神经系统的发育和功能维持非常重要。神经细胞依赖自噬来控制蛋白的质量并清除应激的内质网。正常情况下,内质网自噬对神经细胞有保护作用,其缺陷会导致神经系统疾病。遗传性感觉和自主神经病Ⅰ型(hereditary sensory and autonomic neuropathy type Ⅰ,HSANⅠ)患者存在ATL3突变(Y192C和P338R),导致ATL3与GABARAP的相互作用受阻,引起内质网自噬缺陷[25]。C型尼曼-匹克氏病是一种致命的、进行性的神经退行性疾病,由NPC1突变(突变产物I1061T NPC1)导致的功能缺失引起,可能存在自噬功能障碍[42]。与野生型相比,NPC1突变小鼠FAM134B mRNA水平无显著差异,但FAM134B由内质网转移至自噬体,自噬溶酶体和溶酶体增多,表明尼曼-匹克病虽然自噬流量升高,但可能存在底物降解障碍[43]。在大鼠脊髓神经疼痛模型中,鞘内注射雷帕霉素可促进FAM134B和LC3的表达,增强内质网自噬,缓解内质网应激,减少caspase-3的表达,减轻神经疼痛;相反,鞘内注射自噬抑制剂3-甲基腺嘌呤(3-MA)可使神经疼痛加重。缺乏有效的自噬时,胞质内有毒的蛋白聚集物可导致内质网应激水平升高,细胞凋亡增加[44]。
2.4 内质网自噬与肿瘤 内质网自噬可促进肿瘤的发展。在异柠檬酸脱氢酶1(isocitrate dehydrogenase 1,IDH1)突变的胶质瘤中,2-羟戊二酸(由IDH1突变产生)抑制了胶原-4-辅氨酰羟化酶的活性,导致错误折叠胶原Ⅳ在内质网中积累,触发内质网应激,促进FAM134B的表达及其介导的内质网自噬,导致内质网面积减小。由于内质网是磷脂合成的位点,因此,内质网自噬可导致卵磷脂和脑磷脂合成减少。抑制内质网自噬可恢复IDH突变的神经胶质瘤细胞中的磷脂合成,触发细胞凋亡,抑制肿瘤生长,延长IDH突变的神经胶质瘤小鼠的寿命[45]。
内质网自噬还可促进肿瘤细胞凋亡。对于恶性胶质母细胞瘤,洛哌丁胺可触发转录因子ATF4及其他内质网应激标志物的表达上调,诱导FAM134B和TEX264介导的内质网自噬,引起自噬性细胞死亡[46]。内质网自噬亦可促进肿瘤耐药。布吉他滨通过诱导持续的内质网应激促进结直肠癌细胞凋亡,具有显著的抗癌作用。然而,布吉他滨在介导内质网应激的同时也可促进FAM134B介导的内质网自噬,作为对内质网应激的保护机制。联合使用内质网自噬抑制剂3-MA/氯喹可增强布吉他滨对结直肠癌的抗肿瘤作用[47]。目前,肿瘤领域与内质网自噬相关的研究局限于胶质瘤、胶质母细胞瘤及结肠癌。内质网自噬在不同类型肿瘤中发挥不同的作用,但具体机制有待进一步研究,以实现肿瘤的靶向治疗。
2.5 内质网自噬与微生物感染 内质网自噬与细菌、病毒感染有关系。固有感受器TMEM173/STING可检测内在化的G+细菌产生的c-di-AMP,迅速调动相互依赖的细胞免疫反应,包括内质网应激、哺乳动物雷帕霉素靶蛋白(mTOR)失活和内质网自噬。内质网自噬可恢复巨噬细胞稳态和促进Ⅰ型干扰素反应[48-49]。自噬在病毒感染中发挥双重作用——抗病毒或促进病毒感染,这取决于病毒种类以及病毒的复制阶段。在人类胎儿的神经干细胞中,Zika病毒可抑制Akt-mTOR信号通路,诱导自噬,促进病毒感染,抑制神经发生。自噬也具有抑制病毒复制的作用[50]。虫媒病毒包括Dengue病毒和Zika病毒可利用内质网膜促进自身装配及成熟,内质网膜的降解会限制虫媒病毒的复制。BPIFB3缺失可促进FAM134B依赖的内质网自噬,导致内质网降解增加,抑制Dengue病毒和Zika病毒的复制[51-52]。然而多种虫媒病毒,如Dengue病毒、Zika病毒和West Nile病毒可利用其NS2B3蛋白酶在RHD结构域直接剪切FAM134B,阻止富含内质网及病毒蛋白的自噬体的形成[53]。
3 总结与展望
内质网自噬通过溶酶体降解未折叠蛋白质、错误折叠蛋白质和应激的内质网,维持细胞稳态。目前已发现10种内质网自噬受体,然而,不同应激条件下激活的内质网自噬受体不同,新的内质网自噬受体也有待发现。内质网自噬的生理功能、分子调控机制及其与疾病的关系仍需进一步研究。虽然内质网自噬的研究仍处于新兴阶段,但其参与多种疾病的发生发展,有望成为新的治疗靶点。
