基于2,5-双(三氟甲基)对苯二甲酸配体的锌配合物的合成、晶体结构及性质
2021-12-09杨兴乾魏润芝
夏 祥 王 胜 杨兴乾 樊 荣 魏润芝 刘 峥*, 唐 群*,
(1贵州乌江水电开发有限责任公司思林发电厂,铜仁 565109)
(2桂林理工大学化学与生物工程学院,电磁化学功能物质广西区重点实验室,桂林 541004)
0 引言
当前有机-无机杂化多孔材料得到人们的广泛青睐。依靠不同有机配体的不同性质特点与金属进行自由组配,可以形成一系列具有一定孔结构和特定性质的金属-有机骨架(MOFs)材料[1-5]。MOFs材料是由无机金属和有机配体构筑而成的配位聚合物。改变金属离子、有机配体或者金属离子与有机配体之间的作用力可制备出性质、结构多变的MOFs材料。MOFs材料可广泛应用于催化、分子导体、分子磁体、非线性光学、金属腐蚀与防护等领域。
多元羧酸配体因其多种灵活的键合方式而被广泛用于构筑结构新颖、性质独特的多孔配合物[6-10]。早在 2005年,Rosi等[11]发现采用2,5-二羟基-1,4-对苯二甲酸配体,结合锌离子,可以合成具有一维孔道的MOFs材料。Deng等[12]选择对苯二甲酸类配体,制备出较大孔径的镁基MOFs材料,测试发现,该MOFs材料在包覆绿色荧光蛋白的过程中,结晶度保持稳定。2,5-双(三氟甲基)对苯二甲酸(H2L)属于多元羧酸化合物,易与过渡金属离子配位形成稳定的配位键,易获得多样性的拓扑结构及优异的性能。而且在反应中,H2L的羧基部分或完全失去质子,既可以充当氢键供体又可以充当氢键受体,易形成不同维数的结构,适合作为孔材料。此外,芳香多元羧酸化合物因本身具有共轭双键以及芳香基而具有良好的荧光性质[13-15]。我们以H2L、4,4′-联吡啶(4,4′-bipy)/2,2′-联吡啶(2,2′-bipy)和过渡金属锌盐(ZnCl2)为原料,通过溶剂热法获得了2种新型配合物[Zn(L)2(4,4′-bipy)(H2O)]n(1)和[Zn(L1)(2,2′-bipy)]n(2),并通过单晶X射线衍射、红外光谱等测试方法对其结构进行了表征,同时对其热稳定性及荧光性质进行了研究。
1 实验部分
1.1 试剂与仪器
ZnCl2、H2L、4,4′-bipy、2,2′-bipy、N,N-二甲基甲酰胺(DMF)等均为分析纯试剂,购于上海麦克林试剂有限公司,使用时未经纯化。
所用仪器包括美国Agilent Technologies G8910A单晶衍射仪、用于测定C、N和H元素含量的美国Perkin-Elmer 240Q元素分析仪、日本Shimadzu FTIR-8400型傅里叶红外光谱仪(采用KBr压片)、中国UV-5500PC型双光束紫外可见分光光度计、中国EDXRF型荧光光谱仪、美国SDT-Q600型同步热重/微分热重分析仪。
1.2 配合物的合成
配合物 1:称量 0.027 3 g ZnCl2、0.060 4 g H2L、0.031 2 g 4,4′-bipy溶于含有6 mL DMF和4 mL蒸馏水的烧杯中,用磁力搅拌器搅拌30 min,制得混合溶液。将制得的混合溶液转入25 mL带有聚四氟乙烯内衬的反应釜中,放入烘箱加热至90℃。反应72 h后,以10℃·h-1的速率降温至60℃,再保温2 h,关闭烘箱让其在自然状态下冷却至室温。取出反应釜,有无色透明块状晶体生成,产率为53.36%。元素分析按 C20H12F6N2O5Zn 的计算值(%):C 44.51,N 5.19,H 2.24;实测值(%):C 44.38,N 5.23,H 2.18。
配合物2:称量0.027 3 g ZnCl2、0.060 4 g H2L和0.031 2 g 2,2′-bipy溶于含有6 mL DMF和4 mL蒸馏水的烧杯中,室温下用磁力搅拌器搅拌30 min,制得混合溶液。将混合溶液转入25 mL带有聚四氟乙烯内衬的反应釜中,放入烘箱加热至90℃。反应72 h后,以10℃·h-1的速率降温至60℃,再保温2 h,关闭烘箱让其在自然状态下冷却至室温。取出反应釜,有无色透明块状晶体生成,产率为54.6%。元素分析按 C20H10F6N2O4Zn 的计算值(%):C 46.05,N 5.37,H 1.93;实测值(%):C 46.13,N 5.42,H,1.87。
1.3 晶体结构的测试
选取尺寸大小为0.25 mm×0.23 mm×0.18 mm(1)和 0.21 mm×0.20 mm×0.17 mm(2)的晶 体 ,利用Agilent Technologies G8910A单晶衍射仪,在293 K下采用Mo Kα辐射(λ=0.071 073 nm)、φ-ω扫描的方式来收集晶体的衍射点数据。使用CryAlis pro程序还原原始数据,所有的数据均进行Lp因子校正和经验吸收校正[16]。使用SHLEXS-2014中的直接法解出粗结构,再利用SHLEXL-2014程序对非氢原子坐标及其各向异性温度因子进行全矩阵最小二乘法精修。利用理论加氢获取所有氢原子坐标。有关晶体学数据列于表1中,主要键长和键角列于表2中。
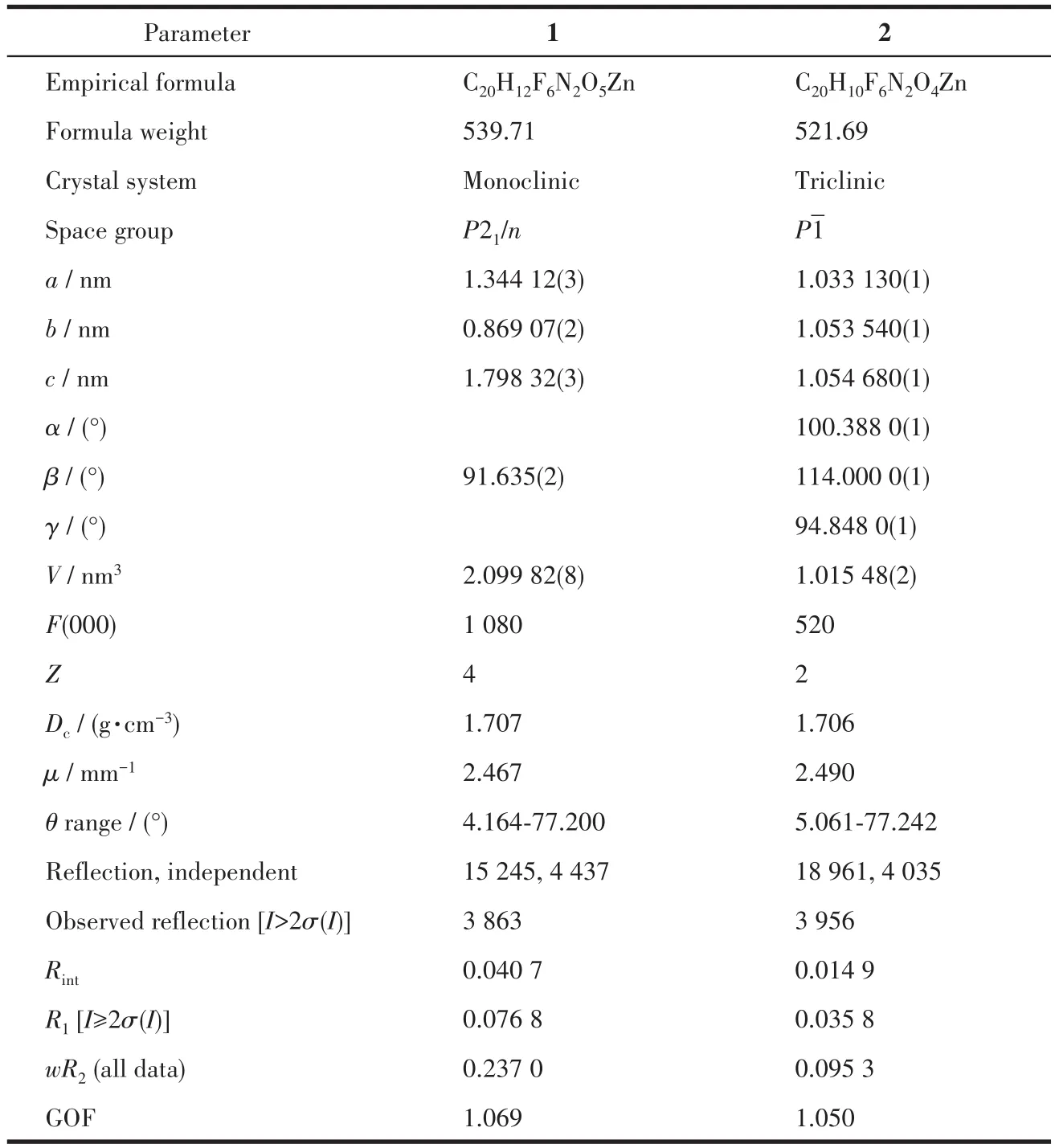
表1 配合物1和2的晶体学数据Table 1 Crystallographic data for complexes 1 and 2

表2 配合物1和2的部分键长(nm)和键角(°)Table 2 Selected bond lengths(nm)and angles(°)for complexes 1 and 2
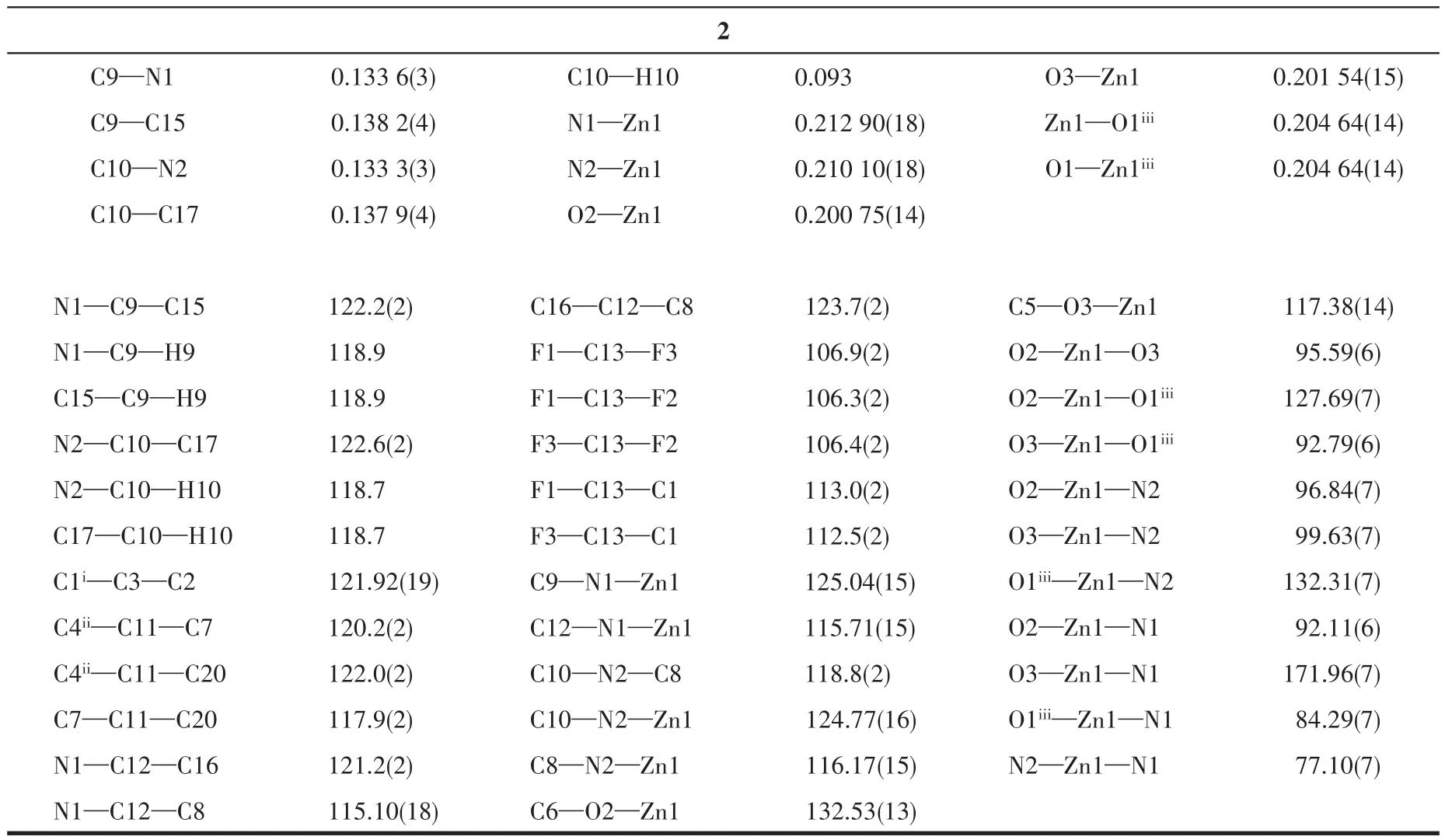
续表2
CCDC:2034067,1;2034068,2。
2 结果与讨论
2.1 配合物的晶体结构
2.1.1 配合物1的晶体结构分析
由图1a可知,配合物1的基本结构单元包含1个Zn(Ⅱ)中心离子、1个主配体L2-、1个辅助配体4,4′-bipy以及1个配位水分子。由图1b可知,在该配合物中,中心金属离子Zn(Ⅱ)分别与配体L2-的3个羧基氧原子、4,4′-bipy的2个氮原子以及1个水分子的氧原子配位,形成六配位结构。配位原子N1、O5、N2iii、O3i与中心原子 Zn(Ⅱ)离子形成的键角之和为359.95°,接近理想的360°,说明这4个配位原子组成了赤道平面,且具有较好的平面性。另2个O2、O4ii位于轴向位置,轴向键角O2—Zn1—O4ii为173.40°,显示出配合物为正八面体构型。

图1 配合物1的晶体结构:(a)30%概率水平的椭球图;(b)多面体图;(c)沿b轴的3D堆积图Fig.1 Crystal structure of complex 1:(a)ellipsoid diagram with 30% probability level;(b)polyhedral diagram;(c)3D stacking diagram along b axis
由表2可知,配位键Zn—O键长处于0.206 1~0.211 2 nm之间,Zn—N键长为0.218 4 nm,键角O—Zn—X(X=O、N)处于 87.96(14)°~177.77(14)°之间,都在正常范围内。由图1c中配合物的b轴方向堆积图可知,配合物是以 Zn2+为金属节点,L2-和 4,4′-bipy作为连接体相互连接,形成无限延伸的二维网状结构,层与层之间通过O5—H5B…O1氢键作用有序堆积形成三维结构。
2.1.2 配合物2的晶体结构分析
如图2a所示,配合物2的每个晶胞含有2个锌配合物单体分子,以其中一个锌配合物单体分子为例,每个不对称单元由1个中心离子Zn2+、1个主配体L2-阴离子以及1个辅助配体2,2′-bipy组成。每个Zn2+离子分别与属于主配体L2-的3个羧基氧原子(O1iii、O2、O3)以及1个辅助配体2,2′-bipy的2个氮原子(N1、N2)配位,形成五配位结构,Zn1iii的配位环境与Zn1一致。由锌、氧和碳3种原子所构成的八边形孔结构孔径为0.474 nm。
图2b显示,每个配位原子N1、O2、O3与中心Zn2+离子形成的键角之和为359.66°,接近理想的360°,说明这3个配位原子组成了赤道平面,且有较好的平面性。配体上的O1iii、N2位于轴向位置,轴向键角 O1iii—Zn1—N2为 132.30°,显示出配合物为三角双锥构型。配位键Zn—O键长处于0.200 75(14)~0.204 64(14)nm之间,Zn—N键长为0.210 10(18)~0.212 90(18)nm。键角 O—Zn—X(X=O、N)处于84.29(7)°~171.96(7)°之间,都在正常的范围内。从图2c中可以看出,配合物2是以Zn2+为金属节点,配体L2-上的每个羧基都通过双齿螯合的方式桥联锌离子,无限连接形成三维网状结构。
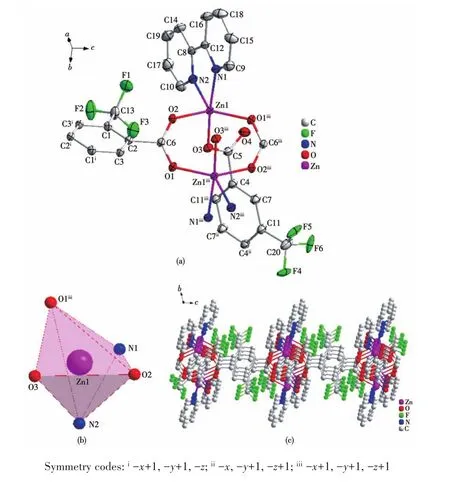
图2 配合物2的晶体结构:(a)30%概率水平的椭球图;(b)多面体图;(c)沿a轴的3D堆积图Fig.2 Crystal structure of complex 2:(a)ellipsoid diagram with 30% probability level;(b)polyhedral diagram;(c)3D stacking diagram along a axis
2.2 配合物1和2的红外光谱
利用日本Shimadzu FTIR-8400型红外光谱仪,以 KBr压片,在 4 000~400 cm-1范围内测定 4,4′-bipy、2,2′-bipy、H2L和配合物1、2的红外光谱(图3)。如图3a所示,H2L在3 430 cm-1处出现羧基中O—H伸缩振动峰,而在配合物1中此峰移动至3 421 cm-1处;H2L在1 717 cm-1处出现羧基的C=O伸缩振动吸收峰,此峰在配合物1中出现在1 659 cm-1处;H2L中934 cm-1处的O—H弯曲振动吸收峰在形成配合物1后移至917 cm-1处。这些都说明锌离子与H2L配体中的羧基配位。4,4′-bipy在1 590 cm-1处显示C=N弯曲振动吸收峰,在配合物1中此峰移动至1 610 cm-1;配合物1在635 cm-1处存在Zn—N吸收峰,这表明4,4′-bipy的氮原子与锌离子配位。
在图3b中,H2L在3 429和3 076 cm-1处显示的羧基中O—H伸缩振动峰,在配合物2的3 495和3 066 cm-1处均再次显示,这表示H2L中的羧基与锌离子进行了配位反应。2,2′-bipy在1 569 cm-1处的C=N伸缩振动峰,在形成配合物2后移至1 621 cm-1处;配合物2在759 cm-1处显示的Zn—N吸收峰说明2,2′-bipy的氮原子与锌离子发生了配位[17-18]。
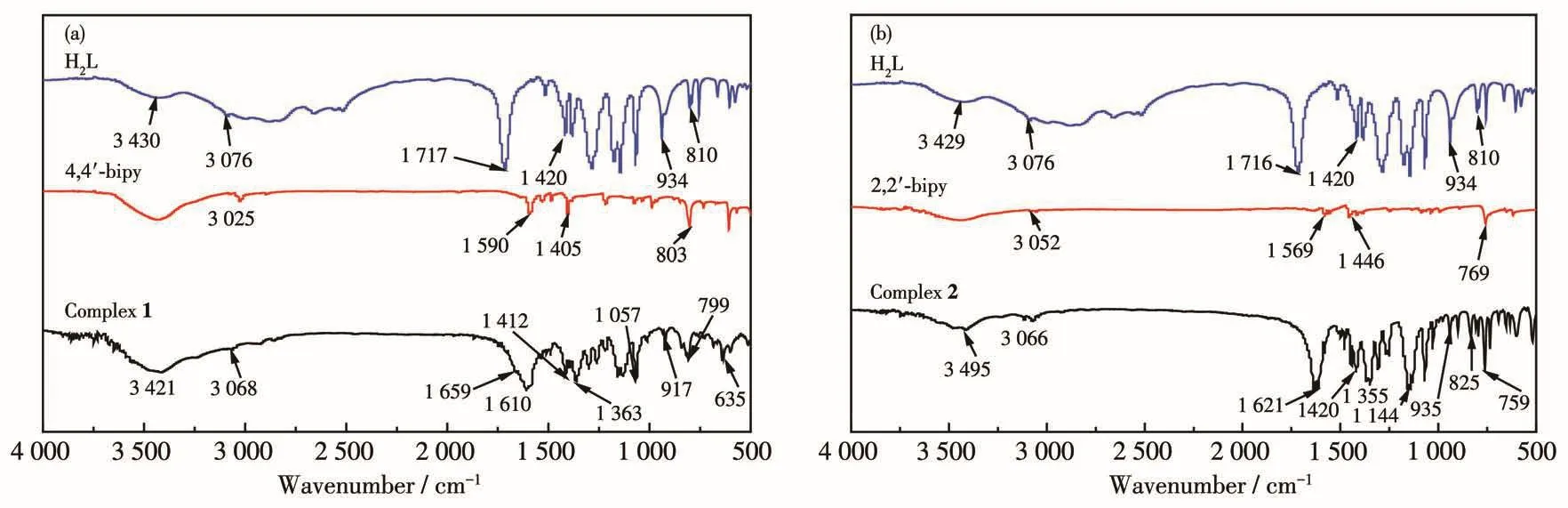
图3 配合物1(a)、2(b)及其配体的红外光谱图Fig.3 IR spectra of complexes 1(a),2(b)and their ligands
2.3 配合物1和2的荧光分析
配合物1、2及其对应配体H2L、4,4′-bipy、2,2′-bipy的荧光发射光谱如图4a、4b所示。所用配体及合成的配合物均含有吡啶环,容易发射荧光,其中配体4,4′-bipy、H2L、配合物1最大发射峰的波长依次为 415、351、348 nm(λex分别为 316、296、333 nm),这归属为π*-π电子跃迁。配合物1、2与配体H2L的发射峰均出现在相近的位置,这表明配合物1、2的荧光主要是配体H2L的发光。配合物2的最大发射波长为349 nm(λex=321 nm),与配体相比,其发射波长发生了红移,这可能是由于金属离子与配体配位后,配合物的最高占据轨道(HOMO)能级降低,导致最低空轨道(LUMO)与HOMO之间的能级差变大[19]。实验结果显示,2种配合物均具有一定的荧光性质。
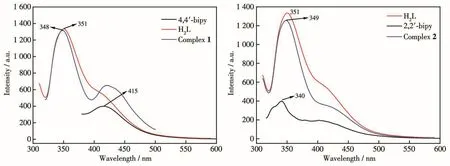
图4 配合物1、2与配体H2L、4,4′-bipy和2,2′-bipy的荧光发射光谱图Fig.4 Fluorescence emission spectra of complexes 1,2 and ligands H2L,4,4′-bipy and 2,2′-bipy
2.4 热重分析
如图5a所示,配合物1在135℃之前比较稳定,未发生分解,在135~440℃范围内发生分解,总的失重率约为83.97%,534℃时热重(TG)曲线趋于平稳,残重为16.03%,残余物可归属为ZnO,理论值为15.07%。如图5b所示,配合物2在239℃之前比较稳定,未发生分解,在239~439℃范围内发生分解,总的失重率约为84.22%,420℃时TG曲线趋于平稳,残重为15.78%,残余物可归属为ZnO,理论值为15.60%。比较可知,配合物1的稳定性比配合物2差。
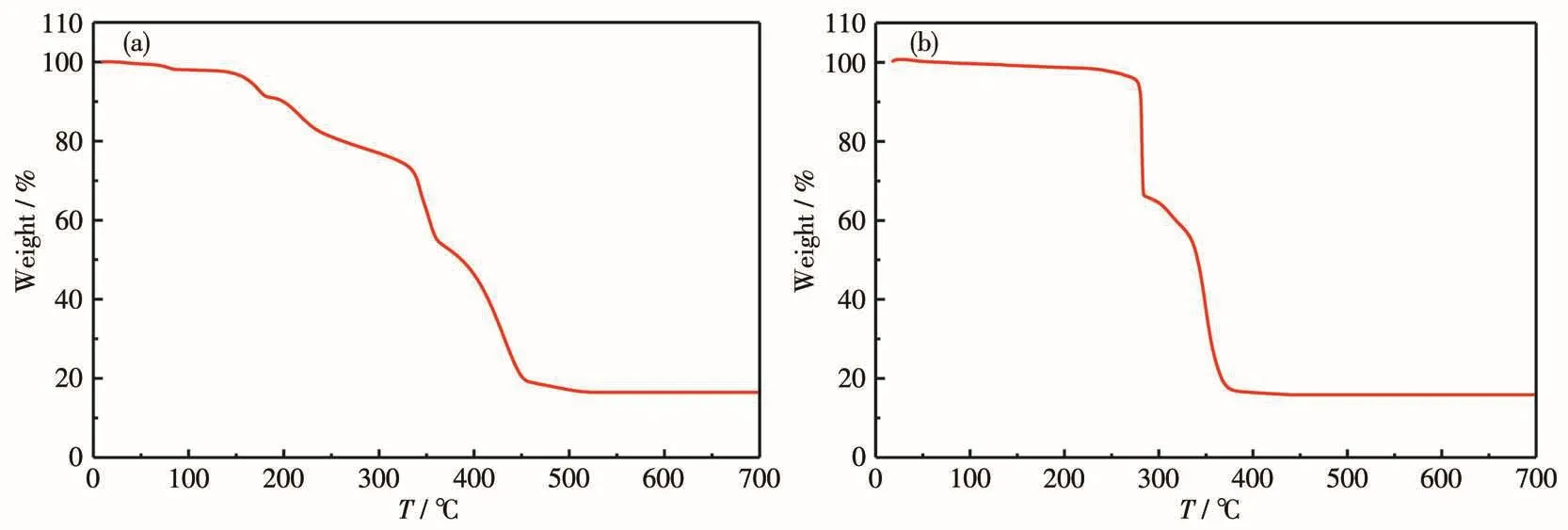
图5 配合物1(a)和2(b)的TG曲线图Fig.5 TG curves of complexes 1(a)and 2(b)
3 结论
以2,5-双(三氟甲基)对苯二甲酸为主配体、4,4′-联吡啶和2,2′-联吡啶为辅配体,与氯化锌通过溶剂热反应,成功合成了2个锌配合物,采用多种分析测试方法对其进行了结构和性质表征,确定合成的物质均为目标产物。配合物1通过层与层之间的分子氢键作用形成三维网状结构,而配合物2是以羧基双齿螯合的方式桥联锌离子,无限连接形成三维网状结构。荧光性质与热稳定性测试结果表明2种配合物均具有良好的荧光性质和热稳定性,配合物1的热稳定性比配合物2差,因此配合物2有望成为潜在的荧光材料。配合物2的中心孔结构有望使其在吸附、负载材料等方面得到应用。
