N掺杂MnO2/碳布复合材料的制备及储锂性能
2021-12-09韩曰朋曾啸雄赵晨鹏谈国强
夏 傲 韩曰朋 曾啸雄 赵晨鹏 谈国强
(陕西科技大学材料科学与工程学院,陕西省无机材料绿色制备与功能化重点实验室,西安 710021)
0 引言
锂离子电池(LIBs)由于能量密度高、循环寿命长、环境友好等特点被广泛应用于电动汽车、便携式电子设备等储能系统中[1-4]。然而,目前商业化的石墨负极由于其较低的理论比容量(372 mAh·g-1)而不能很好地满足于社会需求,因此具有较高理论比容量的过渡金属氧化物逐渐受到关注。其中,MnO2由于具有储量丰富、价格低廉、理论比容量高(1 232 mAh·g-1)和放电平台低等众多优点而被认为是最有潜力的负极材料之一。
MnO2可分为一维隧道结构(α-、β-、γ-MnO2)、二维层状结构(δ-MnO2)和三维网状结构(λ-MnO2)三种类型。其中,δ-MnO2具有的层状结构有利于Li+离子在材料中的脱嵌。然而,MnO2较低的本征电子电导率(10-5~10-6S·cm-1)[5]和充放电过程中的体积效应限制了其可逆比容量及高倍率性能的发挥,因此众多的研究人员将重心放在对MnO2导电性能和结构稳定性的改性上。常用的改性方法包括纳米结构设计、碳材料复合以及掺杂等。其中,掺杂是通过在材料禁带中引入杂质能级从而提高材料导电性的有效手段之一。Zhang等[6]通过水热和煅烧相结合的方法制备了用作Zn离子电池正极的N掺杂MnO2-x/TiC/C材料。N掺杂提高了材料的电化学性能,掺杂后的电极材料在1 A·g-1的电流密度下循环1 000次后仍然保持有172.7 mAh·g-1的可逆比容量,容量保持率为84.7%,然而该电极材料制备工艺过于繁琐。Lin等[7]以聚苯乙烯为模板,聚吡咯为碳源,采用溶液法制备了N掺杂的碳纳米泡,掺杂的杂原子N不仅增大了碳纳米泡的电导率,而且丰富了Li+/Na+离子的活性位点,从而提高了比容量。然而碳材料在高倍率充放电时易出现枝晶锂析出的情况,不适合在大倍率下充放电。He等[8]将MnO2暴露在N2等离子体中制备了用作光催化剂的N掺杂MnO2,能带理论计算证实了N掺杂后材料的导电性有所提高。此外,一些金属离子(如 Mo6+、Zn2+、Fe3+、Al3+等)[9-12]以及非金属离子(B3+、S2-等)[13-14]掺杂 MnO2的报道中也证明了离子掺杂可以提高材料的电化学性能。传统电极由活性材料、导电剂和黏结剂组成,而黏结剂为非电化学活性物质,势必会影响电极材料整体电化学性能的发挥,因此有研究人员开始着眼于无黏结剂电极的制备。Wu等[15]将碳布(CC)作为柔性基底,通过水热法将MnO2纳米片直接生长在CC表面。该材料用作Zn离子电池正极材料时,在1 A·g-1的电流密度下循环300次后仍然保持有263.9 mAh·g-1的可逆比容量。其中,CC为电子传输提供通道,MnO2纳米片提供了较大的比表面积,因而电极具有良好的电化学性能。Zhong等[16]和Xu等[17]分别通过水热法制备了用作超级电容器电极的Ni2+掺杂和Al3+掺杂的MnO2/CC复合材料。此外,还有文献报道了一些将其他过渡金属氧化物(如SnO2、Fe2O3等)[18-19]直接生长在CC表面制备复合材料并用于锂离子电池负极。综合以上文献可以发现,以CC为基底的无黏结剂电极中,CC能够提供良好的电导率和柔韧性[20],在CC上生长的三维网络状纳米材料可以缓解电极材料在充放电过程中的体积效应[21-22],这两方面的协同作用使得电极能够呈现出优良的电化学性能。由于N3-离子(0.171 nm)具有与O2-离子(0.132 nm)相似的半径和不同的价态,在其掺杂进入MnO2晶格中时会引入一定的缺陷,而缺陷的引入有利于材料导电性的提高。因此本实验选择N为掺杂元素,CC为柔性自支撑基底,通过简单的一步水热法制备了N掺杂MnO2/CC无黏结剂负极材料,探究了N掺杂对MnO2/CC的结构、形貌及电化学性能的影响。结果表明在CC柔性基底与N掺杂的共同作用下,N掺杂MnO2/CC复合材料具有较高的可逆比容量和良好的倍率性能。
1 实验部分
1.1 试剂与仪器
实验所用的 KMnO4(纯度 99.0%)、HNO3(纯度99.0%)、无水乙醇(纯度99.0%)、尿素(纯度 99.0%)、丙酮(纯度99.0%)均购于国药集团有限公司;CC购于碳能科技有限公司。
采用D/MAX2200PCX型X射线衍射仪(XRD,日本理学公司)对样品进行物相鉴定,Cu靶Kα为衍射源,波长λ=0.154 06 nm,管电压为40 kV,管电流为40 mA,扫描范围和扫描速度分别为5°≤2θ≤80°和10(°)·min-1。采用SU8100型场发射扫描电镜(SEM,日本HTACHI公司)对样品进行形貌表征,工作电压为30 kV。采用ESCALAB 250Xi型X射线光电子能谱仪(XPS,美国Thermo Fisher Scientific公司)对样品进行元素分析及价态分析,Al Kα(486.6 eV)为激发源,以C1s的电子结合能(284.6 eV)为标准进行误差校正。采用ASAP2460比表面积和孔隙度分析仪(Micromeritics仪器公司,Norcross,GA,美国)测量了样品的N2吸附-脱附曲线,根据77 K时的氮气吸附数据计算比表面积。采用CHI600E型电化学工作站(上海辰华仪器公司)对电池进行循环伏安测试(CV,测试电压为0.01~3.00 V,扫速为0.10 mV·s-1)以及交流阻抗测试(EIS,测试频率范围为0.001~100 kHz)。采用CT2001A型蓝电电池测试系统(LAND,武汉蓝电电子股份有限公司)对电池的循环性能进行测试,电压为0.01~3.00 V。
1.2 实验过程
采用水热法制备N掺杂的MnO2/CC复合材料,合成过程如图1所示。首先对CC进行预处理,将CC裁剪为2 cm×4 cm的长方形长条,然后将裁剪后的CC用丙酮浸泡24 h以去除CC表面的有机物杂质,再用质量分数40%的浓硝酸超声浸泡1 h以引入含氧官能团,之后用去离子水超声清洗3次,每次10 min,60℃下干燥备用。其次,根据本课题组前期的研究成果[23],确定本实验中N的引入量按照原料中N/Mn原子比为10%计算。分别称取0.001 mol的KMnO4(0.158 g)和 0.000 1 mol的尿素(0.006 g)放入40 mL去离子水中,磁力搅拌10 min至均匀溶解,然后将此前驱液倒入聚四氟乙烯内衬中,将第一步中预处理好的CC竖直放入前驱液中,之后在反应釜中120℃水热反应2 h,随后冷却至室温即可取出CC,并用去离子水和乙醇交替冲洗3次,60℃下干燥12 h即得目标产物,记为10%N-MnO2/CC。纯相MnO2/CC的制备过程中不加入CH4N2O,其他实验过程与上述步骤完全一样。

图1 N-MnO2/CC的合成路线Fig.1 Synthetic route of N-MnO2/CC
1.3 电池的组装和测试
以CC为柔性集流体,将活性物质直接生长于CC表面,不再需要涂膜过程。将所制备的电极材料切成直径为16 mm的电极片,其负载量约为1.3 mg·cm-2,以 Li片为对电极,以 1 mol·L-1LiPF6的 DMCEC-DEC(碳酸二甲酯-碳酸乙烯酯-碳酸二乙酯,体积比1∶1∶1)溶液为电解液,在充满氩气的手套箱中进行CR2032型纽扣电池的组装。借助LAND电池测试系统(CT2001A)对所组装的电池进行恒电流充放电测试,电压范围0.01~3.00 V。借助电化学工作站(CHI600E)进行CV测试(0.01~3.00 V)和电化学阻抗(EIS)测试(0.001 Hz~100 kHz)。
2 结果与讨论
2.1 XRD分析
图2为样品的XRD图,其中CC的XRD图与标准卡片PDF No.26-1080对应,在26.6°和44.7°处的衍射峰分别对应于碳的(004)和(102)晶面。当MnO2生长在CC表面之后,其XRD图在12.5°处出现了一个明显的衍射峰,在37.3°处出现了一个微弱的衍射峰,经过与标准卡片(PDF No.80-1098)对比发现这2个衍射峰分别对应于δ-MnO2的(001)和(111)晶面。N掺杂MnO2/CC样品的XRD图中未出现其他杂质峰或含氮化合物的衍射峰,说明产物中没有出现其他含氮的杂质,N元素可能进入了MnO2的晶格。
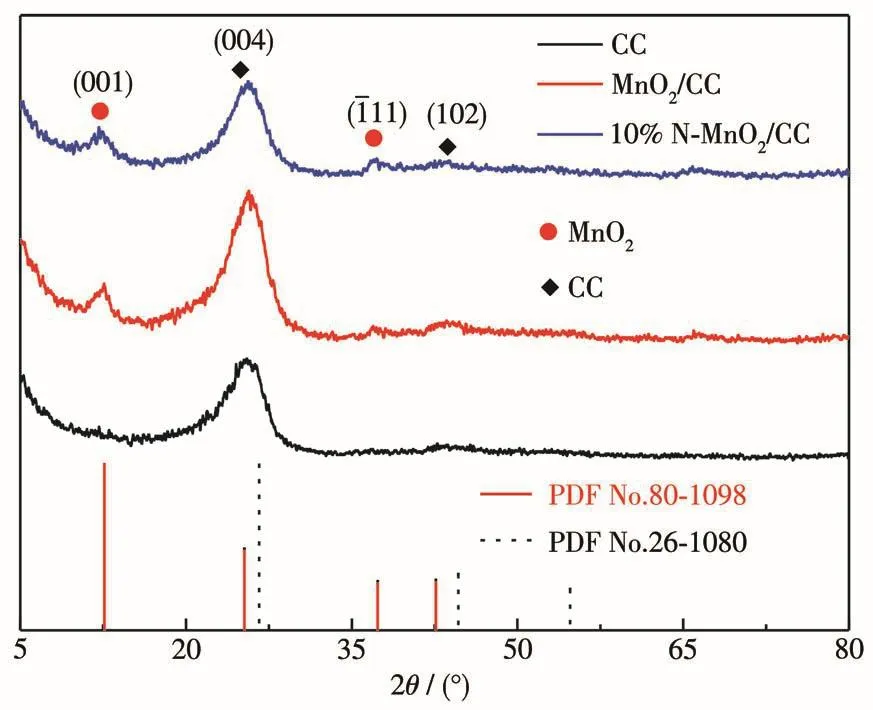
图2 样品的XRD图Fig.2 XRD patterns of the samples
2.2 SEM分析
图3为样品的SEM图片。由图3a、3b可知,纯CC表面较为光滑且无交错褶皱出现,CC纤维的平均直径约为8 μm。生长MnO2的CC表面出现了分散的纳米花球颗粒,花球直径约为500 nm,如图3c和3d所示。图3e为图3d的表面局部放大图,可见在花球颗粒下层生长着由纳米片组装而成的三维网络状MnO2层。N掺杂后,CC表面的花球颗粒分布密度增大,花球直径仍约为 500 nm(图 3f~3g),且花球下面同样有三维网络状MnO2层(图3h),这种特征形貌与其他文献类似[24-25]。三维网络纳米结构有利于电极材料比表面积的提升,从而促进电极材料与电解质充分接触,提供更多的活性位点。MnO2在CC表面的生长分为不同阶段,当水中的MnO4-离子与CC材料接触时,MnO4-离子与C原子发生剧烈反应,MnO4-离子从C原子得到电子,首先在CC表面成核、生长成MnO2纳米片,继而形成致密的纳米片网络状结构。当CC纤维表面布满三维网络状MnO2层时,后续生成的MnO2纳米片以花球颗粒形式分散在外表面[26]。MnO2形成的反应方程式如式1[24]所示。图4为10%N-MnO2/CC的能谱(EDX)图,可以看出N元素掺杂成功且分布均匀。

图3 CC(a、b)、MnO2/CC(c~e)和10%N-MnO2/CC(f~h)的SEM图Fig.3 SEM images of CC(a,b),MnO2/CC(c-e),and 10%N-MnO2/CC(f-h)

图4 10%N-MnO2/CC的EDX元素映射图:C(a);O(b);Mn(c);N(d)Fig.4 EDX elemental mappings of 10%N-MnO2/CC:C(a);O(b);Mn(c);N(d)
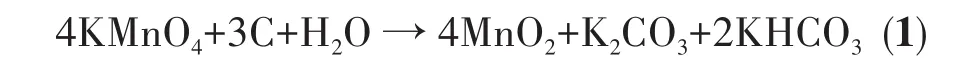
2.3 N2吸附-脱附分析
为了探究N掺杂对样品比表面积的影响,分别对MnO2/CC和10%N-MnO2/CC做了BET(Brunauer-Emmett-Teller)比表面积分析。结果表明,N掺杂前比表面积约为 3 m2·g-1,掺杂后约为 10 m2·g-1,N 掺杂后样品的比表面积是未掺杂样品的3倍多。较大的比表面积有利于电极材料与电解液更加充分地接触,从而增强电极材料的电化学性能。图5a为10%N-MnO2/CC的N2吸附-脱附等温线,根据IUPAC规则可知,该曲线为Ⅳ型等温线,说明其具有介孔结构。从图5b中可以看出其孔径主要分布在4~16 nm之间,而有少量介孔的孔径分布在3.25 nm附近。这种多孔结构有利于促进电解质离子和锂离子的扩散,并且纳米多孔结构可以缓冲充放电过程中MnO2的体积效应,有利于提高电极的倍率性能和循环稳定性。
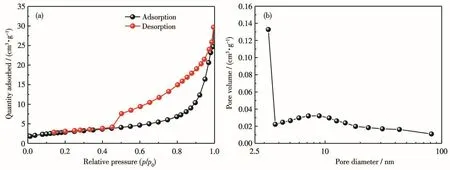
图5 10%N-MnO2/CC的N2吸附-脱附等温线(a)和孔径分布曲线(b)Fig.5 N2adsorption-desorption istherms(a)and pore size distribution curves(b)of 10%N-MnO2/CC
2.4 XPS分析
为了获得样品的表面元素状态,分别对掺杂前后的样品做了XPS分析,如图6所示。从图6a可以看出掺杂前后的样品均主要包含C、Mn、O元素,其中C可能源于空气或者CC基底。由于N为掺杂元素,含量相对较少,因此在全谱图中不易观察。图6b则显示了掺杂前后N1s的高分辨XPS谱图,可见掺杂N后,在399.7 eV(N1)和403.5 eV(N2)结合能处出现了2个明显的峰,这证明N并不以唯一的结合方式存在,其中399.7 eV处的峰对应于掺杂在MnO2晶格间隙的N[8],403.5 eV处的峰对应于N—O[27],其可能以C—O—N的结合方式存在于CC表面。通过对比拟合面积,我们认为大部分的N掺杂在MnO2的晶格间隙。为了探究掺杂N后对Mn和O元素状态的影响,我们对Mn2p以及O1s进行了分峰拟合,如图6c、6d所示。Mn2p分为Mn2p3/2(642.4 eV)和Mn2p1/2(654.1 eV)两个峰,其中Mn2p3/2可以分为642.2和643.5 eV两个峰,Mn2p1/2又可以分为653.7和654.9 eV两个峰。642.2和653.7 eV对应于Mn3+离子,643.5和654.9 eV对应于Mn4+离子,掺杂前nMn3+/nMn4+为1.52,掺杂后为1.93,意味着N的引入有利于Mn3+含量的增加。图6d为掺杂前后O1s的XPS谱图,其中529.9 eV对应于晶格氧(O1),531.1 eV对应于缺陷氧(O2),533.0 eV对应于水分子中的氧(O3),掺杂前nO1/nO2为4.41,掺杂后为3.60,意味着N的引入有利于缺陷氧含量的增加,而Mn3+和缺陷氧含量的增加往往有利于电极材料氧化还原能力的提升,进而提高电化学性能[28-29]。综上所述,N主要掺杂在MnO2的晶格间隙,缺陷形成机理如式2所示,其中Ni‴为间隙氮离子,为氧空位,Mn′Mn代表转化为Mn3+的Mn4+:
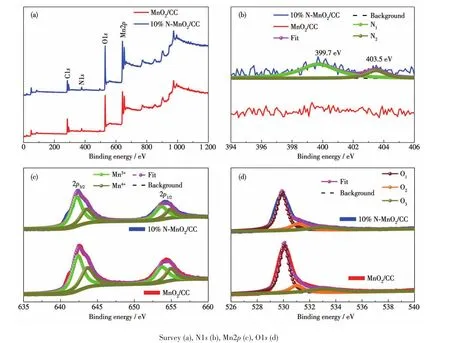
图6 MnO2/CC和10%N-MnO2/CC的XPS谱图Fig.6 XPS spectra of MnO2/CC and 10%N-MnO2/CC
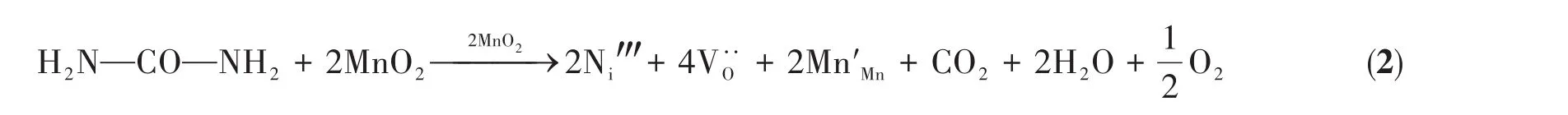
2.5 电化学性能分析
图7a是样品在0.1 mV·s-1扫描速率下的首圈CV曲线。可以看到纯CC主要在低压范围(约0.40 V以下)提供容量,其反应为C+xLi++xe-→LixC(3)[30]。在放电过程中,MnO2/CC和10%N-MnO2/CC都在约0.75 V处出现了一个微弱的阴极峰,这归因于Mn4+→Mn2+的转化,在约0.20 V附近出现了明显的阴极峰,这归因于Mn2+→Mn0的转化。在充电过程中,约1.25 V附近出现了微弱的阳极峰,约2.50 V附近出现了明显的阳极峰,说明金属Mn的氧化分2步进行,分别对应于Mn0→ Mn2+和Mn2+→ Mn4+的转化。图7b为10%N-MnO2/CC前3圈的CV曲线,可以看出曲线吻合程度较高,意味着其具有良好的循环稳定性。图7c为10%N-MnO2/CC的首次充放电曲线,可以看到在放电过程中电压与比容量的变化主要分为3个区域,分别是约0.40 V以上部分(Ⅰ)、约0.20 V的放电平台(Ⅱ)和放电过程最后阶段的倾斜电压(Ⅲ)。在区域Ⅰ中MnO2的电位从3.00 V降到约0.40 V,对应于MnO2转化为Li2MnO2的过程,如式4和5所示,这与图7a中约0.75 V处的阴极峰相对应,而其中的小平台被认为是电解质的分解和SEI膜的形成造成的[31]。在区域Ⅱ中约0.20 V的放电平台对应于Li2MnO2转化为Mn和Li2O的过程,如式6所示,这与图7a中约0.20 V处的阴极峰对应。在放电的最后阶段区域Ⅲ中所释放的容量被认为是通过可逆的电化学反应传递出的,与Mn的氧化还原反应无关[32-33]。在区域Ⅳ的充电过程中,分别在1.25和2.50 V附近出现了2个小平台,二者分别对应于图7a中约为1.25和2.50 V的阳极峰,这2个过程分别发生Mn0→Mn2+和Mn2+→Mn4+的转化。首次充放电后所产生的较大容量损失是电解质的分解和SEI膜的形成造成的。

图7d为3个样品在电流密度0.1 A·g-1下的首次充放电曲线,CC、MnO2/CC和10%N-MnO2/CC的首次充电比容量分别为179.9、605.3和951.9 mAh·g-1,首次放电比容量分别为192.3、762.3和1 194.1 mAh·g-1,库仑效率分别为93.6%、79.4%和79.7%。可见掺杂前后库仑效率基本不变,但掺杂后首次充电比容量较高,这是因为掺杂后电极材料的比表面积大幅提升,提供了更多的嵌锂位点,从而提高了电化学性能。图7e为所有样品在电流密度1 A·g-1下前100圈的循环性能曲线,CC、MnO2/CC和10%N-MnO2/CC的首次充电比容量分别为107.3、447.1、640.3 mAh·g-1,循环100次后可逆比容量分别为94.0、291.3、529.9 mAh·g-1,容量保持率分别为 87.6%、65.2%、82.7%。可以看出10%N-MnO2/CC具有良好的循环稳定性且拥有最高的可逆比容量,远高于商用MnO2[34],这是因为在CC柔性基底上构筑的三维网络状MnO2有助于维持其结构稳定性。图7f为样品的倍率性能曲线,可以看出10%N-MnO2/CC具有优异的倍率性能,其在0.1、0.2、0.5、1、2 A·g-1电流密度下充电比容量分别为948.8、841.7、765.3、657.3、399.9 mAh·g-1,当电流密度重新回到0.1 A·g-1时,充电比容量可达到初始比容量的95.7%(907.9 mAh·g-1)。
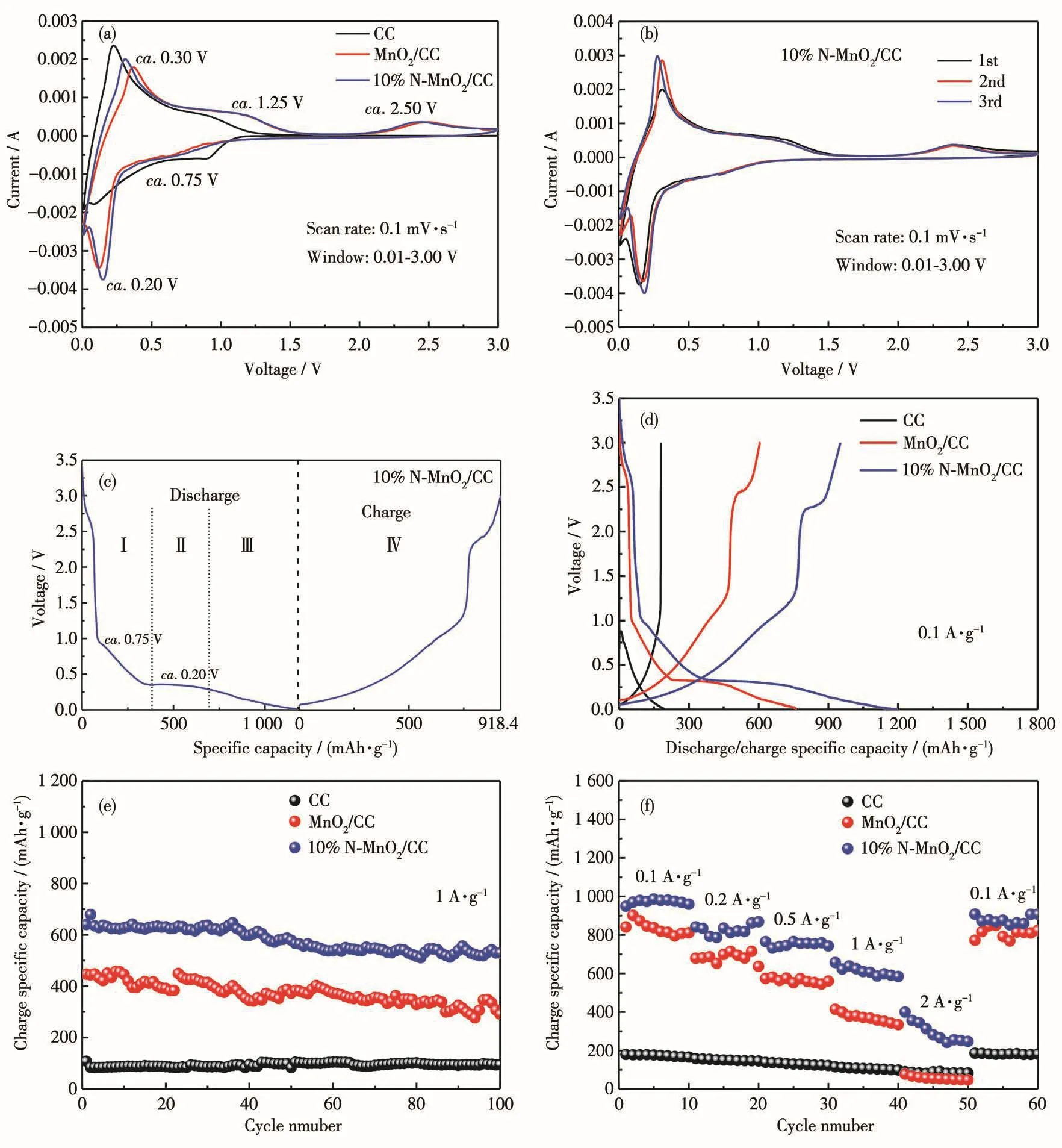
图7 样品的首圈CV曲线(a);10%N-MnO2/CC的前3圈CV曲线(b);10%N-MnO2/CC在首次脱锂/嵌锂周期内的电压曲线(c);样品的首圈充放电曲线(d);样品在电流密度1 A·g-1下的循环性能(e);样品的倍率性能(f)Fig.7 First CV curves of the samples(a);First three CV cycles of 10%N-MnO2/CC(b);Voltage profile of 10%N-MnO2/CC during the first lithiation/delithiation cycle(c);Initial charge/discharge curves of the samples(d);Cycling performance of the samples at 1 A·g-1(e);Rate performance of the samples(f)
从循环测试中可以看出掺N后材料比容量有较大幅度的提升。10%N-MnO2/CC在1 A·g-1电流密度下的首次充电比容量为640.3 mAh·g-1,循环100次后的可逆比容量为529.9 mAh·g-1,容量保持率为82.7%,容量损失~17.3%。为进一步明确电极失效机制,观察了该电极材料在1 A·g-1电流密度下充放电循环100圈后的表面形貌。从图8中可以看出电池经过100圈循环后,电极材料的花球形貌变得模糊且CC纤维表面形貌存在开口破损状态,这可能是造成其容量损失的主要原因。

图8 10%N-MnO2/CC在1 A·g-1下循环100次后不同放大倍率的SEM图Fig.8 SEM images with different magnifications of 10%N-MnO2/CC after 100 cycles at 1 A·g-1
为了进一步探究N掺杂对锂离子在电极材料内部扩散速率的影响,我们对样品进行了EIS测试。图9a为样品在0.1 A·g-1电流密度下循环3圈后的EIS谱图,插图分别为高频区的谱图和等效电路图。在拟合电路中,Rs表示溶液电阻,主要包括电极的固有电阻及离子或电子通过电解液、隔膜和活性材料表面SEI膜的电阻;Rct对应电极的电荷转移电阻;ZW反映了Warburg阻抗,与锂离子在电极内部的扩散相关;CPE对应的是双层电容。拟合数据如表1所示,其中E代表标准误差,所有数据的标准误差均小于10%。可以看出10%N-MnO2/CC具有最小的Rs和Rct,分别为4.11和27.91 Ω,表明具有良好的导电性。通过式7、8[11,35]计算了锂离子扩散系数DLi+,其中R、T、A、n、F、c和σ分别代表理想气体常数、热力学温度、电极表面积、氧化过程中每个分子失去的电子数、法拉第常数、锂离子浓度和阻抗因子。σ与低频区Z′/ω-1/2(Z′为阻抗,ω为频率)有关,Z′-ω-1/2关系图中拟合曲线的斜率即为σ,如图9b所示。DLi+数据如表1所示,10%N-MnO2/CC具有较大的DLi+,这是因为N掺杂后促使电极材料的比表面积增大,改善了电极材料对电解液的浸润性,增加了有效接触面积,同时降低了Rct。图9c为锂离子和电子在充放电过程中的脱嵌与传输示意图。与传统集流体相比,CC为电子快速传输提供了通道,同时,在其表面构筑的三维网络状MnO2增大了电极材料与电解液的接触面积,为锂离子的脱嵌提供了更多的活性位点。

图9 样品在0.1 A·g-1下循环3次后的EIS谱图(a);Z′和ω-1/2在低频区的关系曲线(b);10%N-MnO2/CC负极中Li+和e-的传输路径模型(c)Fig.9 EIS spectra of the samples after three cycles at 0.1 A·g-1(a);Relationship between Z′and ω-1/2at low frequency range(b);Schematic diagram of transport path model of lithium ions and electrons with 10%N-MnO2/CC anode(c)
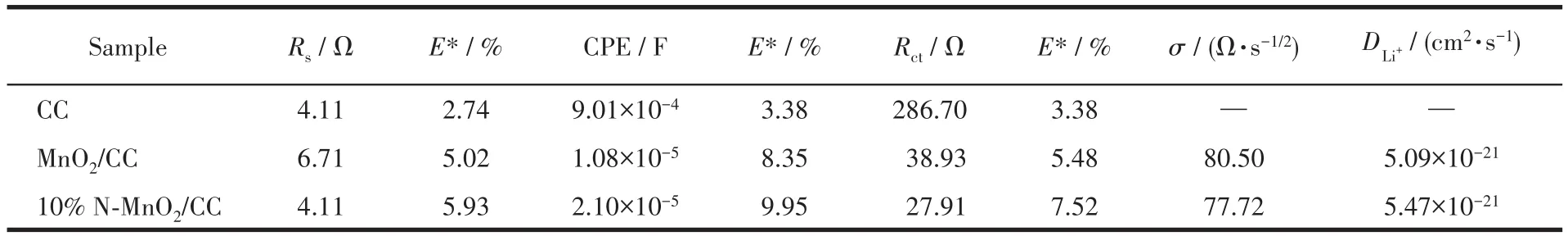
表1 样品在0.1 A·g-1下循环3次后的EIS拟合数据Table 1 EIS fitting parameters for the samples after three cycles at 0.1 A·g-1
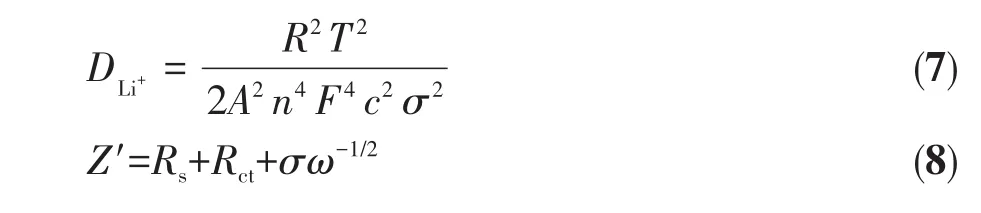
3 结论
以CC为柔性基底,通过水热法制备了三维网络状MnO2/CC和10% N-MnO2/CC负极材料,借助XRD、SEM、XPS等表征手段确定了产物的组成和形貌。电化学性能测试表明10% N-MnO2/CC具有良好的电化学性能,在0.1 A·g-1电流密度下具有907.9 mAh·g-1的可逆比容量,在1 A·g-1的大电流密度下首次充电比容量为640.3 mAh·g-1,循环100次后仍有初始比容量的82.7%(529.5 mAh·g-1)。该材料优异的电化学性能主要归功于以下3点:(1)CC为电子的快速传输提供了路径,在CC表面构筑的多孔网络状MnO2有利于缓解锂离子脱嵌过程中的体积效应;(2)N的掺杂使得CC表面MnO2花球颗粒增多,增加了电极材料比表面积,有利于电解质与电极材料的充分接触,为锂离子提供了更多的反应场所;(3)N的掺杂增加了电极材料中Mn3+和氧空位的含量,使得电极材料的氧化还原能力和导电性增强。
