气相色谱法测定盐酸羟胺肟化反应液中环己酮、环己酮肟和硝基环己烷的含量
2021-11-29王瑞菲唐晓婵陈贵军冯维春
邢 伶,王瑞菲,唐晓婵,陈贵军,岳 涛,冯维春
(青岛科技大学 山东省水相有机合成及高效清洁分离工程技术研究中心,济南 250014)
盐酸羟胺是一种重要的化工原料,广泛应用于石油化工、医药、农药、电化学、橡胶等行业[1]。以环己酮、氨水、过氧化氢溶液、盐酸为原料,经过酮的氨氧化(肟化反应)和酮肟水解制备得盐酸羟胺,其因具有清洁环保、原料价廉、毒性低、安全系数高等优势成为近年来的研究热点[2-4]。在制备盐酸羟胺的工艺中,环己酮作为起始原料不断被循环利用,生成的环己酮肟经盐酸水解生成盐酸羟胺,其中副产物硝基环己烷随着反应的进行不断累积,当达到一定含量时会影响催化剂的活性和工艺安全。
目前,环己酮的测定方法主要有气相色谱法[5-8],利用环己酮显色反应的分光光度法[9-10]以及高效液相色谱法[11]。环己酮肟多采用间接法测定,如在酸性介质中,将环己酮肟水解成羟胺,利用羟胺的显色反应,通过分光光度法对其进行测定[12-13];也有文献采用高效液相色谱法[14-15]以及气相色谱法[16]测定环己酮肟的含量。硝基环己烷的测定方法有气相色谱-热导检测器法[17]、气相色谱-氢火焰离子化检测器法[18]。关于同时测定盐酸羟胺反应液中环己酮、环己酮肟和硝基环己烷含量的研究未见报道。
为简化分析过程、缩短分析时间,本工作建立了气相色谱法同时测定盐酸羟胺肟化反应液中环己酮、环己酮肟和硝基环己烷的方法,用于跟踪酮和酮肟的转化率以及监测硝基环己烷的累积程度,可为工艺人员调整优化盐酸羟胺工艺流程提供技术指导。
1 试验部分
1.1 仪器与试剂
Trace 1300型气相色谱仪,配氢火焰离子化检测器;SQP型电子天平(感量0.000 1 g)。
环戊酮肟内标溶液:称取环戊酮肟1.0 g,用无水乙醇溶解并定容至100 mL容量瓶中,配制成10 g·L-1环戊酮肟内标溶液。
混合标准溶液A:称取环己酮、环己酮肟、硝基环己烷各1.0 g,置于100 mL容量瓶中,用无水乙醇稀释至刻度,配制成10 g·L-1的混合标准溶液A。
混合标准溶液B:移取混合标准溶液A 1.0 mL于100 mL容量瓶中,用无水乙醇稀释至刻度,配制100 mg·L-1的混合标准溶液B。
混合标准溶液系列:取6个100 mL容量瓶,各加入5.0 mL环戊酮肟内标溶液,再依次移取1.0,10.0 mL混合标准溶液 B和1.0,2.0,5.0,10.0 mL混合标准溶液A,用无水乙醇稀释至刻度,配制成1.0,10.0,100.0,200.0,500.0,1 000.0 mg·L-1的 混合标准溶液系列。
环己酮、环戊酮肟的纯度不小于99%,环己酮肟的纯度不小于98%,硝基环己烷的纯度不小于97%;无水乙醇为色谱纯。
1.2 仪器工作条件
Kromat KB-624色谱柱(30 m×0.32 mm,1.8μm);载气为氮气;气化室温度250℃;流量2.0 mL·min-1;进样量1μL;分流比为100∶1;检测器温度为250℃。柱升温程序:初始温度为80℃,保持3 min;以30℃·min-1的速率升温至200℃,保持6 min。
1.3 试验方法
称取盐酸羟胺肟化反应液0.1 g于100 mL容量瓶中,加入5.0 mL环戊酮肟内标溶液,用无水乙醇稀释至刻度,配制成样品溶液,按仪器工作条件进行测定。
2 结果与讨论
2.1 色谱条件的选择
2.1.1 色谱柱
试验考察了Kromat KB-624弱极性色谱柱和Wonda CAP WAX极性色谱柱对待测物分离效果的影响。结果显示,两种色谱柱均能分离待测物和内标物,当使用Kromat KB-624色谱柱时,色谱峰对称性更好,相邻色谱峰的分离度均大于1.5,保留时间适中,故试验选择Kromat KB-624色谱柱进行后续分析。
2.1.2 柱温和柱流量
选择柱温时,既要保证待测物与相邻组分完全分离,又要保证各组分全部出峰,并且分析时间越短越好,以提高分析效率。试验考察了等温方式和程序升温方式对待测物分离效果的影响。结果显示:由于各待测物的极性和沸点存在差异,等温方式不能获得较好的分离效果,因此试验采用程序升温方式来分离待测物,柱升温程序见1.2节。同时试验考察了柱流量为1.0,1.5,2.0,2.5,3.0 mL·min-1时对各待测物分离效果的影响。结果显示:当柱流量为1.0,1.5 mL·min-1时,环己酮肟的色谱峰拖尾;当柱流量为2.0 mL·min-1时,各待测物的峰形和分离度均较好。因此,试验选择柱流量为2.0 mL·min-1。
2.1.3 分流比
反应液中各组分含量随着反应的进行不断变化,导致各待测物含量跨度较大,因此需要选择合适的分流比,防止样品过载或检测器灵敏度过低。试验考察了分流比为50∶1,75∶1,100∶1,125∶1时各待测物的色谱行为。结果表明,随着分流比的增大,各待测组分的色谱峰峰形与分离度均有改善,但是当分流比过大时,各待测物的响应信号过低。综合考虑,试验选择分流比为100∶1。
2.2 定量方法和内标物
由于不同反应液中各待测物含量差别较大,且同一反应液中3种待测物含量的差别也较大,使用外标法测定高含量的待测物时,进样量和气相色谱系统的微小变化均可能导致峰面积出现较大变化。并且,试验发现使用单点内标时,每次得到的校正因子的变化较大,导致测定高含量待测物的结果准确度和重复性较差。因此,试验选择内标法定量,以消除进样体积和系统条件变化对测定结果的影响。
一般选择待测样品中不存在、不与样品中各组分发生化学反应、保留时间与待测组分接近且能完全分离的化合物为内标物,通常选择化学结构与待测组分相似的同系物或异构体。根据上述原则,试验考察了丙酮、丙酮肟和环戊酮肟为内标物时对3种待测物分离效果和峰形的影响。结果显示:由于丙酮沸点较低,出峰时间较快,与溶剂峰(无水乙醇)的分离度小于1.5,不能完全分离;以丙酮肟为内标物时,丙酮肟与样品溶液中其他未知杂质峰分离不佳,干扰测定;以环戊酮肟为内标物时,环戊酮肟与相邻待测物色谱峰均能达到基线分离,且内标物峰形良好,出峰时间适宜。因此,试验选择环戊酮肟作为内标物。
2.3 色谱行为
取适量无水乙醇、200.0 mg·L-1的混合标准溶液与样品溶液,按照优化的试验条件进行测定,所得色谱图见图1。
由图1可知:混合标准溶液和样品溶液中环己酮、环戊酮肟、环己酮肟、硝基环己烷色谱峰与相邻组分的分离度均大于2.0,峰形良好,并且溶剂无水乙醇不干扰样品的测定。

图1 色谱图Fig.1 Chromatograms
2.4 标准曲线、检出限和测定下限
按照试验方法对混合标准溶液系列进行测定,以各目标物的质量浓度为横坐标,其对应的峰面积与内标物峰面积的比值为纵坐标绘制标准曲线。结果表明:3种化合物标准曲线的线性范围均为1.0~1 000.0 mg·L-1,相关系数均大于0.999 0。具体的线性参数见表1。

表1 线性参数Tab.1 Linearity parameters
以3倍信噪比(S/N)和10倍信噪比计算检出限(3S/N)和测定下限(10S/N),得到环己酮、环己酮肟和硝基环己烷的检出限均为0.02 mg·L-1,测定下限均为0.08 mg·L-1。
2.5 精密度试验
称取盐酸羟胺反应液0.1 g,平行制备6份样品溶液,按照试验方法进行测定,所得环己酮、环己酮肟和硝基环己烷的测定值及其相对标准偏差(RSD)见表2。

表2 精密度试验结果(n=6)Tab.2 Results of test for precision(n=6)
由表2可知:环己酮、环己酮肟、硝基环己烷测定值的 RSD分别为0.89%,0.82%,2.8%。
2.6 回收试验
称取盐酸羟胺反应液0.05 g,对样品进行低、中、高3个浓度水平的加标回收试验,每个浓度水平平行测定6次,计算回收率和测定值的RSD,结果见表3。
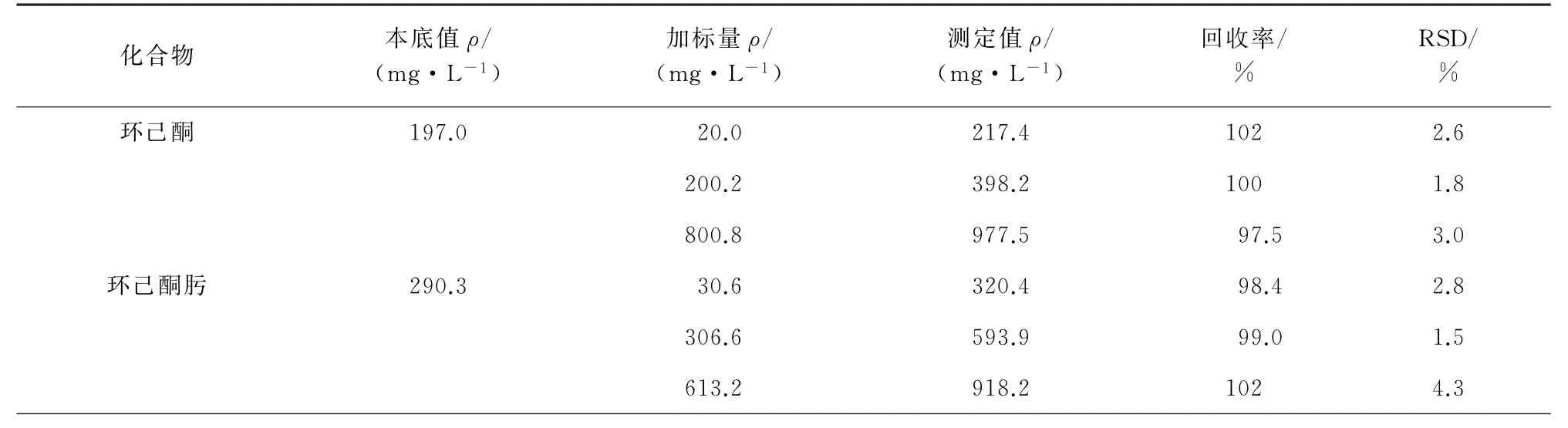
表3 回收试验结果(n=6)Tab.3 Results of test for recovery(n=6)

表3(续)
由表3可知:环己酮、环己酮肟和硝基环己烷的回收率为97.5%~102%,测定值的RSD为1.5%~4.5%。
2.7 样品分析
取8批盐酸羟胺肟化反应液S1~S8,按照试验方法进行测定,结果见表4。

表4 样品分析结果Tab.4 Analytical results of samples %
本工作通过优化色谱柱、柱温、流量、分流比、内标物等条件,建立了气相色谱法同时测定盐酸羟胺肟化反应液中环己酮、环己酮肟和硝基环己烷含量的方法。该方法具有准确、快速、稳定的优点,满足测定要求,适用于盐酸羟胺肟化工艺中环己酮、环己酮肟和硝基环己烷的监测。
