快速测定唑尼沙胺血药浓度的高效液相色谱法
2021-11-19张忠新任方龙李瑞光郭庆合贺志安
王 侠,张忠新,任方龙,李瑞光,郭庆合,贺志安
(1.新乡医学院医学检验学院,河南省免疫与靶向药物重点实验室,河南省分子诊断与医学检验技术协同创新中心,河南 新乡 453003;2.新乡医学院第三附属医院检验科,河南 新乡 453003;3.郑州爱微迪医学检验实验室有限公司,河南 郑州 450019)
唑尼沙胺是一种新型广谱抗癫痫药物,可有效治疗包括部分性发作、继发性全身发作等多种类型的癫痫。有研究发现,唑尼沙胺可明显改善帕金森病患者的临床症状[1-2],自2009年以来唑尼沙胺被推荐用于帕金森病的补充治疗[3]。
唑尼沙胺在体内吸收迅速,最大吸收时间2~6 h,生物利用度>95%。唑尼沙胺不会诱导自身的代谢,也不会抑制细胞色素P450酶系统;但苯妥英钠、卡马西平和巴比妥酸盐会加速唑尼沙胺的代谢并降低其半衰期[4];有研究显示,唑尼沙胺可抑制苯妥英钠在体内的代谢[5]。唑尼沙胺可引起患者发生不良事件,最常见的有嗜睡(17%)、头昏(13%)、厌食(13%)和头痛(10%)[6]。唑尼沙胺还会影响某些癫痫患者延迟的单词回忆、口语流利性,且具有剂量依赖性[7]。据报道,服用唑尼沙胺易致代谢性酸中毒[8],因此,美国食品和药物管理局要求抗癫痫药物唑尼沙胺的生产厂商在其产品标签上增加有关该药物存在代谢性酸中毒风险的警告。另外,唑尼沙胺在不同性别、给药次数的患者间药物峰浓度存在差异,女性患者给药后血药浓度明显高于男性,多次给药后血药浓度明显大于单次给药[9]。综上所述,尽管唑尼沙胺利用度较高,但其药物毒副作用可引发不良事件,与其他药物共用时存在相互影响。因此,有必要对唑尼沙胺血药浓度进行监测,尤其对于女性,避免由于药物浓度过大而引起不良反应的增加,实现个体化用药。
本研究参照国内外相关方法,依据本实验室条件,利用高效液相色谱(high performance liquid chromatography,HPLC)平台建立了测定人血清中唑尼沙胺浓度的方法,并依据《中国药典》2020 年版第四部通则中“生物样品定量分析方法验证指导原则”[10]对该方法进行验证,旨在为临床上检测唑尼沙胺血药浓度及预防毒副作用的发生等提供依据。
1 材料与方法
1.1 试剂与仪器纯度 99.9%唑尼沙胺对照品、纯度 99.3%苯代三聚氰胺对照品购自梯希爱(上海)化成工业发展有限公司,分析纯醋酸铅购自天津市科密欧化学试剂有限公司,色谱纯甲醇、乙腈购自美国Fisher公司;LC-20A HPLC仪、AUW220D十万分之一分析天平购自日本岛津公司,Centrifuge 5427R高速离心机购自德国Centrifuge Eppedorf公司,ST70-2恒温振动器购自美国Thermo公司,PAL-CAXXBIOM2超纯水机购自美国Pall corporation公司,OF1旋涡混合器购自上海琪特分析仪器有限公司,ACE UltraCore 2.5 SuperC18色谱柱(100.00 mm×3.00 mm)购自英国ACE公司。
1.2 唑尼沙胺标液、质控液和内标工作溶液配制
1.2.1 唑尼沙胺标液的配制称取唑尼沙胺标准品粉末125.68 mg,将其溶解于100 mL甲醇中,即可获得1.256 8 g·L-1的唑尼沙胺储备液。已知唑尼沙胺对照品纯度为99.9%,唑尼沙胺储备液真实质量浓度为1.255 5 g·L-1。将唑尼沙胺储备液使用甲醇逐级对半稀释,连同唑尼沙胺储备液一起,得到L6-L1系列浓度的标液,质量浓度分别为1 255.5、627.75、313.88、156.94、78.47、39.23 mg·L-1。
1.2.2 内标工作溶液的配制称取苯代三聚氰胺对照品粉末216.09 mg,将其溶解于100 mL甲醇中,即可获得质量浓度为2.160 9 g·L-1的苯代三聚氰胺储备液。吸取250 μL储备液,用乙腈定容至100 mL,即可获得质量浓度为5.40 g·L-1的内标工作溶液。
1.2.3 不同浓度质控液的配制取300 μL唑尼沙胺储备液加入100 μL甲醇混匀即为高浓度质控液;取200 μL唑尼沙胺储备液加入200 μL甲醇混匀即为中浓度质控液;取100 μL唑尼沙胺储备液加入300 μL甲醇混匀,再从中取出100 μL加入甲醇300 μL 混匀即为低浓度质控液;从低浓度质控液中取200 μL加入200 μL甲醇混匀即为定量下限浓度质控液。
1.3 色谱条件ACE UltraCore 2.5 SuperC18(100.00 mm×3.00 mm)色谱柱;流动相:A相为超纯水,B相为纯甲醇,梯度洗脱(0.01~5.00 min,18%~50%甲醇;5.00~5.50 min,50%~18%甲醇;5.50~7.50 min,18%甲醇),流速0.6 mL·min-1,进样量20 μL,柱温30 ℃,检测波长240 nm。
1.4 观察指标
1.4.1 方法的选择性分别取6份未服用唑尼沙胺健康志愿者的血清为空白血清考察方法的选择性,样品处理方法:取100 μL空白血清,加入10 μL的400 g·L-1的醋酸铅水溶液,再加入300 μL的乙腈,充分涡旋混匀1 min后放置10 min,25 ℃下12 000 r·min-1离心10 min。取100 μL上清液移至已加入100 μL超纯水的96孔进样板内,25 ℃下500 r·min-1离心10 min,最后吸样20 μL进行HPLC定量分析。以峰面积响应值衡量干扰组分。接受标准:干扰组分响应低于分析物定量下限响应的20%,并低于内标响应的5%。
1.4.2 样品残留评价残留的考察是通过高浓度样品分析后即刻分析空白样品来进行。高浓度样品制备方法:取唑尼沙胺储备液200 μL加入50 μL甲醇混匀,从中取10 μL加入到90 μL空白血清中,加入10 μL 400 g·L-1的醋酸铅水溶液,再加入300 μL内标工作溶液,平行制备5份高浓度样品。空白样品制备方法:取100 μL空白血清,加入10 μL 400 g·L-1的醋酸铅水溶液,再加入300 μL乙腈。各样品充分涡旋混匀1 min后放置10 min,25 ℃下12 000 r·min-1离心10 min。各取100 μL上清液移至已加入100 μL超纯水的96孔进样板内,25 ℃下500 r·min-1离心10 min,最后吸样20 μL进行HPLC定量分析。
1.4.3 线性评价精确吸取空白血清90 μL于1.5 mL EP管内,分别加入L6-L1标液10 μL、400 g·L-1醋酸铅水溶液10 μL及内标工作溶液300 μL,充分涡旋混匀1 min后放置10 min,25 ℃下12 000 r·min-1离心10 min。取100 μL上清液移至已加入100 μL超纯水的96孔进样板内,25 ℃下500 r·min-1离心10 min,最后吸样20 μL进行HPLC定量分析。每份标本重复检测3次,评价仪器对唑尼沙胺分析物的响应。通过已知浓度的分析物与仪器检测分析物与内标面积并计算比值建立标准曲线。
1.4.4 准确度与精密度评价精确吸取空白血清 90 μL于1.5 mL EP管内,分别加入各浓度质控液10 μL、400 g·L-1醋酸铅水溶液10 μL及 内标300 μL,充分涡旋混匀1 min后放置10 min,25 ℃下12 000 r·min-1离心10 min。取100 μL上清液移至已加入100 μL超纯水的96孔进样板内,25 ℃下500 r·min-1离心10 min,最后吸样20 μL进行HPLC定量分析。每个浓度平行5份。3个浓度质控液每天测5次计算批内精密度、准确度,连续测定3 d计算批间精密度、准确度。准确度均值应在质控样品标示值的±15%范围内,定量下限应在标示值的±20%范围内。批内、批间精密度相对标准偏差(relative standard deviation,RSD)不超过15%,定量下限RSD不超过20%[10]。
1.4.5 样品稳定性评价将配制好的唑尼沙胺储备液加入到空白血清中,配成低质量浓度(7.85 mg·L-1)和高质量浓度(100.44 mg·L-1)血清质控样品。取90 μL空白血清加入1.5 mL EP管,分别加入各浓度质控液10 μL,充分涡旋混匀1 min后放置10 min,25 ℃下12 000 r·min-1离心10 min。取100 μL上清液移至已加入100 μL超纯水的96孔进样板内,25 ℃下500 r·min-1离心10 min,于室温条件下放置24 h后吸样20 μL进行HPLC定量分析,评价处理后样本室温放置24 h的稳定性。将90 μL空白血清加入1.5 mL EP管内,分别加入10 μL各浓度质控液,混匀并密封,于室温存放24 h后充分涡旋混匀1 min,放置10 min,25 ℃下12 000 r·min-1离心10 min。取100 μL上清液移至已加入100 μL超纯水的96孔进样板内,25 ℃下500 r·min-1离心 10 min,最后吸样20 μL进行HPLC定量分析。评价未经处理样品室温放置24 h的稳定性情况。取 1.5 mL EP管,加入90 μL空白血清,分别加入10 μL 各浓度质控液,混匀并密封,于4 ℃冰箱中存放1周后充分涡旋混匀1 min,放置10 min,25 ℃下12 000 r·min-1离心10 min。取100 μL上清液移至已加入100 μL超纯水的96孔进样板内,25 ℃下500 r·min-1离心10 min,最后吸样20 μL进行HPLC定量分析,评价未经处理样品4 ℃存放1周的稳定性。
1.5 统计学处理应用SPSS 19.0软件进行统计学描述。
2 结果
2.1 方法的选择性结果见图1。唑尼沙胺、苯代三聚氰胺的保留时间分别为3.63、4.78 min,唑尼沙胺及内标处干扰符合要求。

A:空白血清;B:苯代三聚氰胺;C:唑尼沙胺+苯代三聚氰胺;1:苯代三聚氰胺;2:唑尼沙胺。
2.2 样品残留评价各分析物在空白血清中的残留与定量下限浓度峰面积比值均<20%,内标在空白中的残留与定量下限浓度峰面积比值均<5%。残留量均符合要求。
2.3 线性考察结果以唑尼沙胺质量浓度为横坐标x、唑尼沙胺峰面积与内标峰面积之比为纵坐标y,进行线性回归得唑尼沙胺的标准曲线方程为y=0.024 480 4+0.011 778 5(R2=0.999)。线性考察结果,唑尼沙胺在3.92~125.55mg·L-1的质量浓度范围内线性良好。
2.4 准确度与精密度考察结果见表1。除唑尼沙胺在定量下限第2批的批内准确度为76.73%外,其余定量下限及低中高浓度的精密度与准确度结果均符合要求。

表1 准确度与精密度试验结果
2.5 样品稳定性结果见表2。经处理血清质控品于室温存放24 h,其准确度稳定;未经处理血清质控品样品于室温下存放24 h、4 ℃下放置1周,准确度稳定。
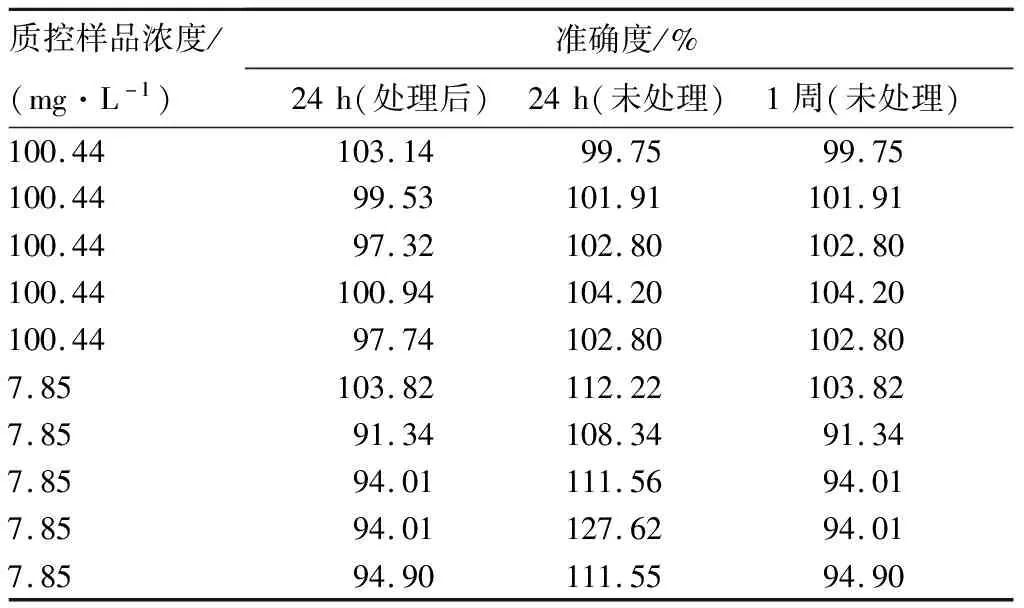
表2 稳定性考察结果
3 讨论
近期国外有学者采用乳胶颗粒增强比浊免疫测定(latex particles enhanced turbidimetric immunoassay,LTIA)方法进行唑尼沙胺浓度的测定,结果显示,在高浓度和低浓度下,HPLC与LTIA法定量的浓度之间无差异,但LTIA法可能无法确定血清水平维持在目标范围以下的患者血清唑尼沙胺的确切浓度,或者可能无法在治疗药物监测中用于确认依从性,当LTIA方法显示质量浓度低于5 mg·L-1时,必须使用HPLC法确认其确切的水平[11]。因此,HPLC依然是比较理想的唑尼沙胺检测方法。
本实验室利用HPLC平台,建立了测定人血清中唑尼沙胺浓度的方法,并参照《中国药典》2020 年版第四部通则中“9012 生物样品定量分析方法验证指导原则”,对该方法进行了验证。苯代三聚氰胺与唑尼沙胺结构相似,分子量接近,其性质稳定、保留时间适当,与唑尼沙胺峰无干扰,适用于唑尼沙胺色谱分析的内标。经筛选,唑尼沙胺与苯代三聚氰胺在240 nm紫外吸收波长下响应最强,无内源性干扰。
本研究所用HPLC色谱为二元泵系统,具备梯度系统条件。笔者曾尝试等度洗脱,峰宽和峰形皆不理想;采用梯度洗脱可改善峰宽和峰形拖尾,提高分析方法的灵敏度。因此实验过程采用梯度洗脱法。据报道流动相多采用乙腈-磷酸二氢钠溶液[12]、甲醇-水[13]、甲醇-乙腈-三氟乙酸水溶液[5]等,本实验中流动相选择A相为超纯水和B相为纯甲醇(18%~50%),方法优化中尝试过不同组成的流动相(加改性剂如甲酸、加乙酸铵等,不同比例甲醇水以及乙腈水等),但分离效果均不理想。使用纯水-甲醇作为流动相的分离效果最佳且保留时间重复性高、峰宽小且对称性好,并且与宣贵达等[13]所采用的甲醇水(8020)相比,降低了有机溶剂用量、试剂成本,可延长色谱柱寿命。
HPLC测定唑尼沙胺血药浓度对于蛋白沉淀有乙腈[11]、甲醇[5]、硫酸锌甲醇沉淀蛋白法[12],其操作繁琐或耗时。本研究采用两步法,即乙腈和醋酸铅沉淀蛋白法。将内标溶液与乙腈混合后获得内标工作溶液,此过程同时添加了内标和蛋白沉淀剂,使操作步骤简化,且相同体积的乙腈沉淀蛋白效果明显优于甲醇,因此,经少量(300 μL)内标工作液处理后,蛋白沉淀效果即可满足实验需求。醋酸铅作为沉淀蛋白沉淀剂,不仅可降低有机溶剂的使用量,提高分析方法的灵敏度,而且相比其他重金属溶剂沉淀蛋白,醋酸铅的污染轻,且与蛋白形成复合物可作为医疗垃圾处理。笔者曾尝试硫酸铵等盐溶液辅助沉淀蛋白,但效果均不理想。因此,加入醋酸铅后蛋白沉淀更加完全,干扰少,离心后即可上机检测,完成1个标本的检测仅需7.5 min,较韦斯军等[12]的9.1 min更迅速。另外本法样本用量少,使用超纯水稀释后进行分析,降低了样品的挥发,同时改善了峰形。因此,本方法具有简单、快速、结果准确的特点,可实现高通量样本的快速测定。
综上所述,本研究建立了测定唑尼沙胺血药浓度的HPLC分析方法,其干扰少、低残留,准确度高、精密度好,线性关系良好,满足临床检测需求。本法样本适应性好,稳定性高,分析物处理后室温可放置24 h,未处理样品可稳定存放于室温24 h、4 ℃ 1周。因此,在规定时间内可保证检测结果能准确地反映患者采样时的药物浓度,从而满足实验室因标本量大需分批检测的要求。本法操作简便,所需血清仅100 μL,可小样本送检,适宜唑尼沙胺药物浓度常规监测,便于样本集中处理。
