自身免疫性胶质纤维酸性蛋白星形细胞病的脑膜脊膜病变研究*
2021-10-30姚海燕江丽红李惠璐刘思高聪龙友明杨新光
姚海燕,江丽红,李惠璐,刘思,高聪,龙友明,杨新光
广州医科大学附属第二医院神经内科、神经科学研究所(广东广州 510260)
慢性或亚急性激素反应性非血管炎性自身免疫炎症性脑膜脑炎(chronic or subacute cortico steroid-responsive nonvasculitic autoimmune inflammatory meningoencephalitis)或非血管炎性自身免疫炎症性脑膜脑炎(nonvasculitic autoimmune inflammatory meningoencephalitis)[1]是自身免疫性胶质纤维酸性蛋白星形细胞病(glial fibrillary acidic protein astrocytopathy,GFAP-A)的前身。2016年梅奥医院的Lennon团队对该类疾病进行报道和统一命名[2]。GFAP-A常累及脑膜、脑、脊髓和视神经等不同的神经部位,它的临床表现包括发热、头痛、脑病、脊髓炎、视力异常,特异性的诊断标记物为GFAP抗体[3-6]。脑的核磁共振(MRI)显示GFAP-A的病灶部位包括大脑白质、基底节、下丘脑、脑干、小脑、脑膜、脑室甚至颅骨[3-6]。最特异的MRI改变是垂直于脑室的脑白质“血管样放射强化”,见于40%~50%的患者[4,6]。GFAP蛋白在软脑膜上和其边缘组织表达较强,发生GFAP-A时,活化的T细胞可能首先靶向脑膜位置诱发炎症反应[7]。头痛往往是GFAP-A最常见的症状之一,提示了脑膜炎症病变。但是,目前尚无对GFAP-A脑膜和脊膜影像学表现开展的针对性研究。笔者团队在2013年开始GFAP-A相关研究,报道了系列GFAP-A患者[8]的临床表现。在本研究中,我们探讨、分析28例GFAP-A患者脑膜和脊膜病变的MRI特点,旨在提高临床和放射学医师对GFAP-A的认识与诊治。
1 资料与方法
1.1 研究对象 纳入从2013年3月至2019年6月在广州医科大学附属第二医院住院且检测脑脊液GFAP-IgG阳性的患者28例,28例患者均在本院完成头颅MRI的平扫和增强(25例同时完成脊髓MRI平扫和增强)。对照组为同期住院的27例血清水通道蛋白4抗体(aquaporin-4,AQP4)阳性的视神经脊髓炎谱系疾病(neuromyelitis optica spectrum disorder,NMOSD)患者,均完成头颅和脊髓MRI平扫和增强。本研究为回顾性研究,收集的资料包括年龄、性别、临床表现、脑与脊髓MRI特征、实验室检测和诊治预后。研究得到广州医科大学附属第二医院伦理委员会批准(批号:2017-hs-23),并在事先告知患者其标本可能会被纳入相关的研究工作的情况下获得患者的知情同意。
1.2 GFAP-IgG和AQP4-IgG抗体检测 GFAP-IgG通过基于组织和(或)基于细胞方法检测脑脊液,具体见本研究组的以往报道[4]。AQP4-IgG通过基于细胞法检测血清,详见研究组以往的研究报道[4]。
1.3 统计学方法 全部数据通过SPSS 11.0统计软件,计数资料通过Fisher确切概率法统计,计量资料通过非参数检验Mann-Whitney-Wilcoxon检测,P<0.05 为差异有统计学意义。
2 结果
2.1 一般情况 28例GFAP-A组中,男15例,女13例,中位发病年龄44(21~77)岁,28例患者主要临床症状有头痛13例(46.4%)、发热16例(57.1%)、脊髓炎12例(42.9%)、视觉异常11例(39.3%)、行为异常8例(28.6%)、共济失调7例(25.0%)、意识障碍4例(14.3%)、癫痫发作3例(10.7%)、运动障碍2例(7.1%)、认知障碍3例(10.7%)和其他症状7例(25.0%)(表1)。
27例NMOSD 组中,男2例,女25例,中位发病年龄42(16~72)岁,全部AQP4-IgG阳性,无伴随其他神经相关抗体(表1)。
2.2 脑、脊膜异常的资料 GFAP-A组中,MRI增强下15例(53.6%)有脑膜和(或)脊膜的异常信号,其中脑膜异常8例(28.6%),脊膜异常3例(10.7%),脑膜与脊膜同时异常4例(14.3%)。8例患者有病理学资料(7例行立体定位活检术和1例因脑膜瘤行手术切除),其中2例显示脑膜增厚,慢性炎症改变(图1~3)。本研究对比了GFAP-A与NMOSD患者的多项资料,结果显示性别比例、发热、头痛、行为异常、视觉异常、MRI放射性血管样强化、脊髓异常长节段脊髓炎、脑膜和(或)脊膜异常差异均有统计学意义(P<0.05)。比较的结果见表1。
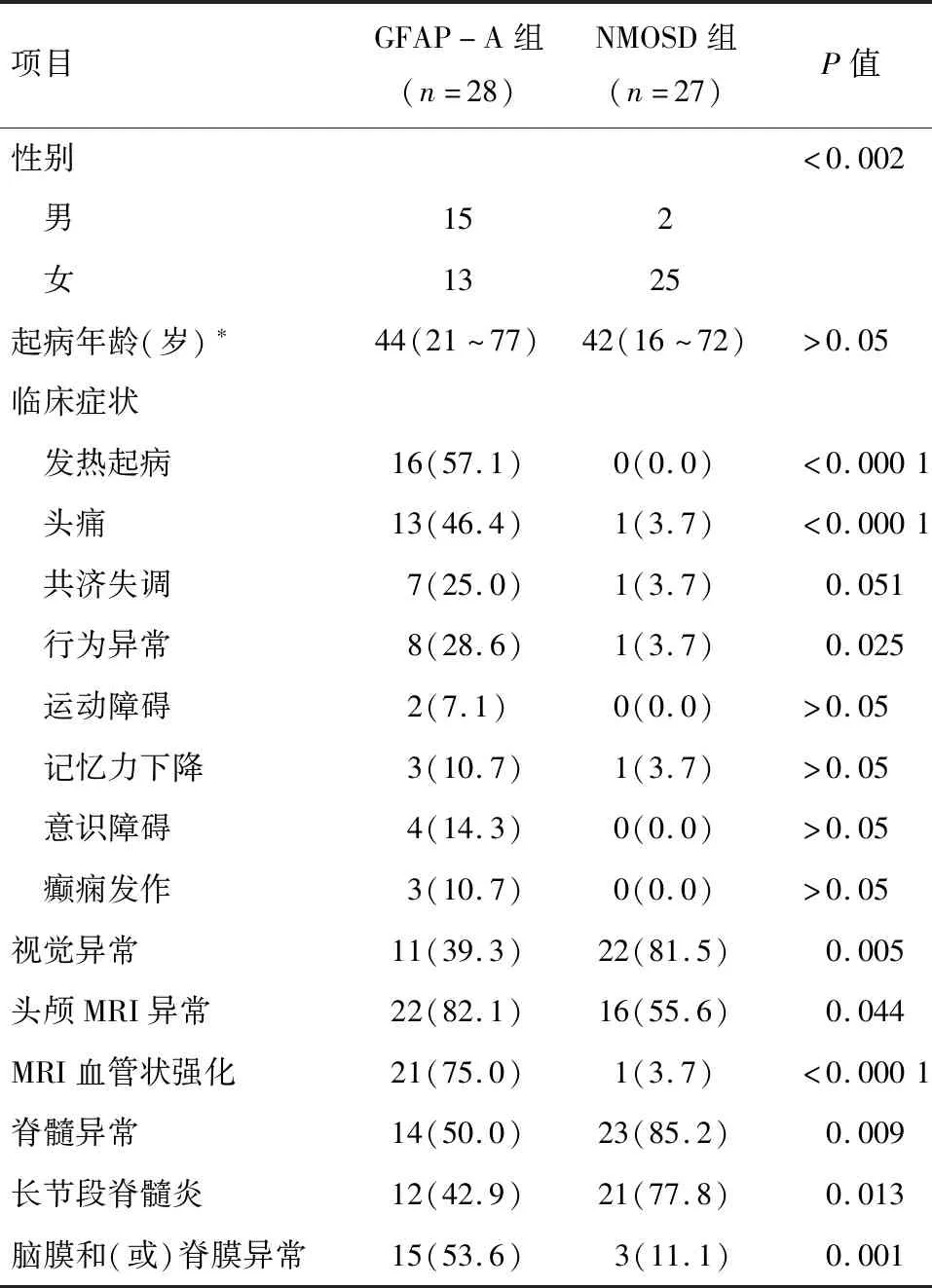
表1 GFAP-A与NMOSD的资料对比 例(%)
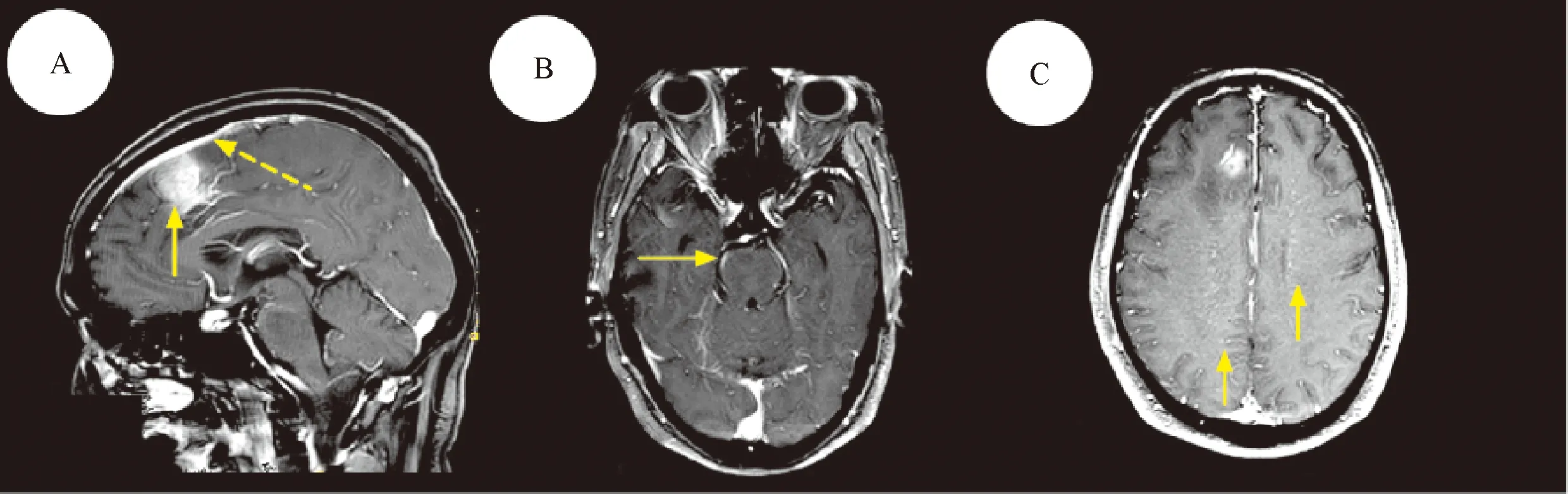
注:患者为40岁亚急性起病的男性,病史约1月余,临床上表现为发热、不自主运动和精神行为异常。A:额部占位并强化(实线箭头),脑膜部位明显增厚(虚线箭头);B:MRI增强显示脑桥部位软脑膜强化(箭头);C:头颅MRI增强显示远离肿物处的放射状血管强化,符合GFAP-A影像学特征(箭头)图1 患者头颅MRI

注:A:苏木精-伊红染色显示占位部位为脑膜瘤合并出血(×400);B:苏木精-伊红染色示软脑膜血管周围炎症(箭头)(×100);C:CD8阳性的T细胞(箭头)(×400)图2 脑活检组织的病理学特征
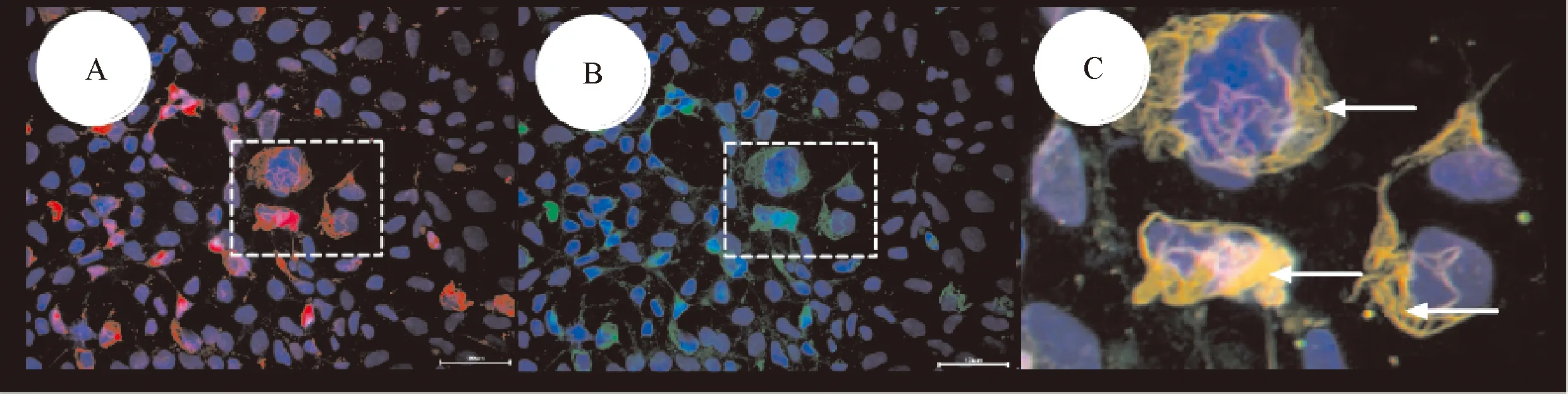
注:A:患者脑脊液荧光显像(×100);B:GFAP-IgG荧光显像(×100);C:荧光双标显像(×400)图3 细胞法检测脑脊液抗体强阳性
3 讨论
自身免疫性星形细胞病是近年来提出一个新的概念,包括了NMOSD和GFAP-A两种类型,它们的诊断标记物分别为AQP4-IgG和GFAP-IgG,靶抗原均分布在星形细胞上,尤其在终突的软脑膜上。因此,理论上软脑膜会成为免疫攻击的靶点。本研究组和其他研究组以往在NMOSD病例中均发现MRI上软脑膜的异常[9-10]。
研究显示GFAP-A患者的MRI显示出多发病灶,累及的部位包括大脑白质、基底节、下丘脑、脑干、小脑等。最特异的MRI改变是垂直于脑室的脑白质“血管样放射强化”,见于40%~50%的患者[6]。GFAP-A典型表型为脑膜脑炎与脑膜脑脊髓炎,其突出的临床症状为头痛,往往是提示脑膜受累的证据。我们的前期研究亦发现个别患者出现脑膜、脑室甚至颅骨异常[4]。病理学显示为脑膜炎与小血管周围炎[7]。在本研究中,我们重新回顾了曾在本院完成头颅MRI平扫和增强的GFAP-A患者,结果发现脑膜脊膜的异常出现于超过50%的患者,甚至部分患者并没有头痛症状仍然合并脑膜的强化和增厚。Mayo医院的报道大约22%的GFAP-A患者出现MRI下的软脑膜的异常信号[6]。因此,我们的发现率明显高于该报道,具体原因不清楚,可能与样本量偏少造成数字波动有关,或者纳入的表型差异等,将来大样本的统计有助于减少这种差异。我们首次报道了2例活检的患者,病理结果亦支持脑膜受累增厚和慢性炎症[7]。同样,我们对比了另一种星型细胞疾病的类型——NMOSD,结果发现GFAP-A脑膜和(或)脊膜病灶发现率远高于NMOSD,可能与两种疾病的发病机制差异有关。NMOSD中,AQP4-IgG具有致病作用,AQP4-IgG阳性的NMOSD特点是病灶内AQP4抗原表达缺失、补体沉积、星形细胞减少[11]。它的机制是AQP4抗体与表达于星形细胞终突的AQP4结合,激活补体,其下游通路涉及兴奋性氨基酸转运体-2 内吞,最后导致髓鞘脱失及组织坏死。GFAP是星形细胞胞内表达的中间丝蛋白,GFAP抗体并不容易与其接触反应产生体液免疫反应。在我们的病理研究中,脑膜炎症位置并没有补体沉积,提示了这种非体液免疫反应特点。动物模型研究提示GFAP特异的T细胞参与了GFAP星形细胞病发病[12],我们的病理学研究显示活化的T细胞,包括CD4和CD8阳性的T细胞出现在脑实质和脑膜部位,支持细胞免疫反应的存在。因此,本结果提示了软脑膜作为血液和中枢交接处更容易受到T细胞的攻击,MRI强化是增强剂从破坏的血脑屏障渗出的结果。随着治疗的好转,血脑屏障迅速修复,强化亦迅速消失。
尽管GFAP-A的命名和重新定义已经有3年余,但是至今尚未见有统一的诊断标准。我们曾推荐患者临床表现为脑膜脑炎、脑炎、脑膜炎、脑膜脑脊髓炎、脊髓炎而MRI显示特征性的血管样增强需考虑GFAP-A可能[13]。我们认为,有上述临床表现的患者,需要关注其MRI的脑膜脊膜病变。由于脑膜脊膜异常在MRI下显示具有一定局限性,临床或者放射学医生可能容易忽视,因此,需提高对GFAP-A影像学特点的认识。
