HPLC-DAD 双波长法同时测定乌蕨中原儿茶酸、原儿茶醛、牡荆苷的含量
2021-09-15胡燕珍张媛媛吴先昊虞金宝
胡燕珍,黄 斌,张媛媛,陈 乐,吴先昊,虞金宝
(江西省中医药研究院,江西 南昌 330046)
乌蕨Stenoloma chusanum(Linn.) Ching为鳞始蕨科乌蕨属多年生草本植物,又称乌韭、大叶金花草、野鸡尾、金花草等[1],生于林下或灌丛中湿地,广泛分布于长江以南各地,且北达陕西南部[2],是一种民间常用中草药,具有清热解毒、活血利湿、止血生肌之效。现代药理学研究表明,乌蕨药理作用广泛、药用价值高,具有抗菌、抗炎、保肝、抗肿瘤、降血糖等多种生物活性[3-5]。乌蕨中含有大量黄酮类、酚酸类及挥发油等化学成分[6]。其地上部分总黄酮含量高达30%以上[7],该类成分具有较强的抗氧化和清除自由基能力,可有效抑制脂质过氧化,其抗氧化能力与乌蕨中总黄酮含量呈正相关[8];其中黄酮类成分牡荆苷对食管癌细胞特别是EC-109细胞增殖有明显抑制作用,能够诱导癌细胞凋亡[8]。而酚酸类化合物的药理活性也较广泛,具有抗菌、消炎、抗病毒 、抗氧化、降血脂、抗凝血等作用[9],其中原儿茶酸、原儿茶醛具有明显的抗菌活性[10]、抗炎[11]、抗蛋白纤维化[12]、抗氧化应激[13]等作用。经查阅文献发现,目前多以牡荆苷、原儿茶酸、原儿茶醛等单一或单一类成分含量来控制乌蕨药材的质量[14-15]。由于中药成分的复杂性及药理作用的多样性,而以单个或单一类指标成分的含量来评价药材质量的优劣,未能真实、全面地反映出药材的内在质量。基于此,本研究采用HPLC技术,建立同时测定乌蕨中原儿茶酸、原儿茶醛、牡荆苷三种指标性成分含量的方法,以期为后续乌蕨药材质量标准的建立提供可靠参考。
1 仪器与材料
1.1 仪器
Agilent 1260型高效液相色谱仪(美国安捷伦科技 公 司);Dikmatech Diamonsil Plus C18(250 mm ×4.6 mm,5 μm; 柱 号:2509215,Dikma Technologies);MS105Du型十万分之一天平(瑞士梅特勒—托利多公司);KQ-250DB型数控超声波清洗器(江苏省昆山市超声仪器有限公司);103B型200 g高速中药粉碎机(浙江省瑞安市永历制药机械有限公司)。
1.2 材料
1.2.1 试药 原儿茶酸(批号:110809-201906,纯度:97.7%,中国食品药品检定研究院);原儿茶醛(批号:110810-201608,纯度:99.3%,中国食品药品检定研究院);牡荆苷(批号:111687-201704,纯度:94.9%,中国食品药品检定研究院)。乙腈和甲醇为色谱纯,水为娃哈哈纯净水,其它试剂均为分析纯。
1.2.2 药材 实验材料于2019年至2020年分别采集于江西各地,共计11个批次样品,经江西省中医药研究院虞金宝研究员鉴定为鳞始蕨科植物乌蕨Stenoloma chusanum(Linn.) Ching的全草,药材来源信息,见表1。
表1 样品信息Tab. 1 Sample information
2 方法与结果
2.1 色谱条件
色谱柱:Dikmatech Diamonsil Plus C18(250 mm ×4.6 mm,5 μm);流动相:乙腈-0.4%磷酸溶液梯度洗脱;检测波长:260 nm(原儿茶酸、原儿茶醛)、340 nm(牡荆苷);流速:1.0 mL/min;柱温:30 ℃;进样量:10 μL,见表2。

表2 梯度洗脱程序Tab. 2 Gradient elution procedure
在该色谱条件下,样品中3种活性成分(原儿茶酸、原儿茶醛、牡荆苷)的峰与其它非被检测峰能够达到基线分离。理论塔板数按原儿茶酸峰计算不低于6 000,R>1.5。混合对照品溶液、供试品溶液色谱图,见图 1。
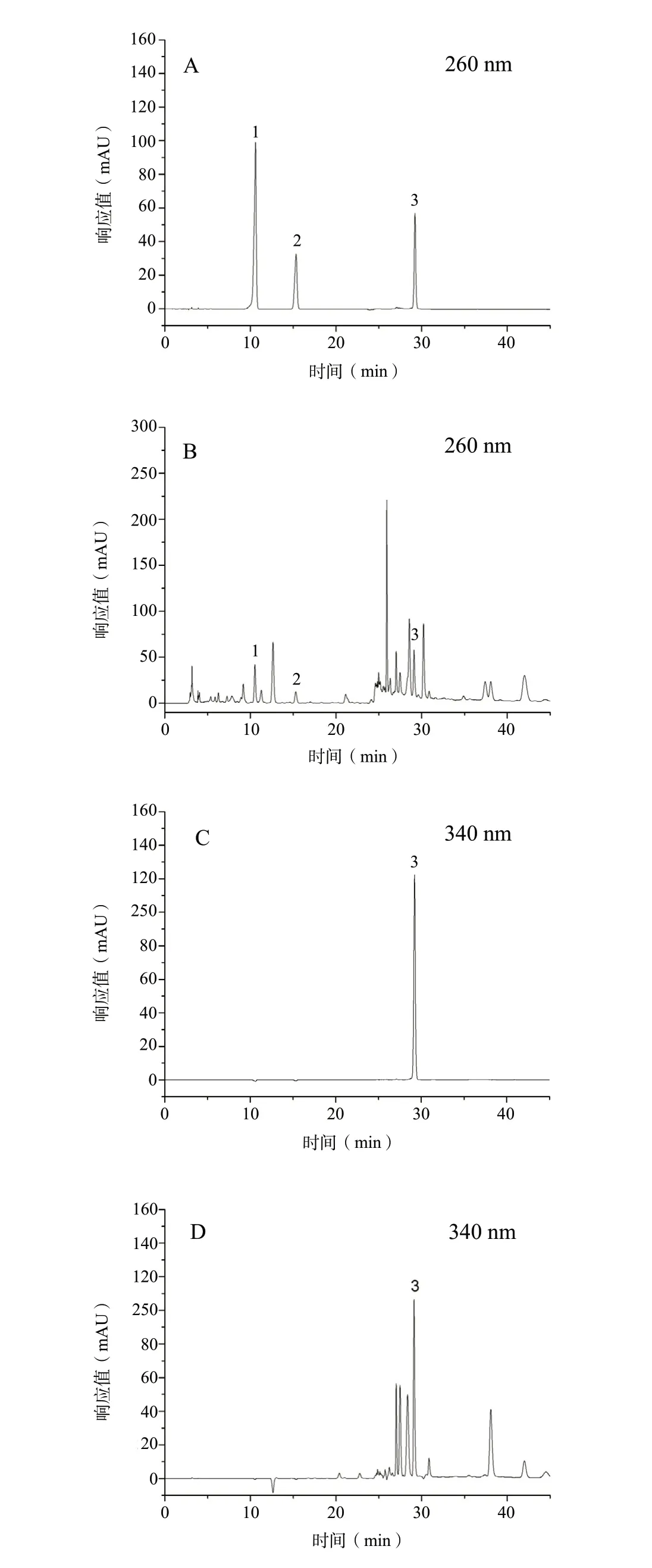
图1 混合对照品溶液(A、C)和供试品溶液(B、D)的HPLC色谱图Fig.1 HPLC chromatograms of mixed control(A/C)and sample(B/D)
2.2 对照品溶液的制备
分别精密称取原儿茶酸、原儿茶醛、牡荆苷对照品适量,加70%甲醇制成各成分对照品母液(原儿茶酸0.251 3 mg/mL 、原儿茶醛0.282 0 mg/mL、牡荆苷0.238 4 mg/mL)。再精密吸取原儿茶酸对照品母液2 mL、原儿茶醛对照品母液2 mL、牡荆苷对照品母液6 mL置于10 mL容量瓶中,加70%甲醇稀释至刻度,即得原儿茶酸、原儿茶醛、牡荆苷的混合对照品储备溶液(质量浓度分别为50.26 μg/mL、56.40 μg/mL、143.04 μg/mL)。
2.3 供试品溶液的制备
取各批次乌蕨药材粉末(过三号筛)约1 g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25 mL,称定重量,超声(功率:250 W,频率:40 kHz)60 min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,经 0.22 μm 微孔滤膜过滤,即得。
2.4 线性关系考察
精密吸取“2.2”项下各对照品母液,用70%甲醇逐步稀释,得到系列混合对照品溶液。原儿茶酸浓度分别为50.26、40.21、15.08、12.06、9.05、5.026 μg/mL;原儿茶醛浓度分别为56.40、45.12、16.92、13.54、10.15、5.640 μg/mL;牡荆苷浓度分别为143.04、114.43、95.36、76.29、57.22、14.304 μg/mL。精密吸取上述各溶液10 μL,按“2.1”项下色谱条件进行测定,记录峰面积,以对照品的进样量为横坐标X,峰面积为纵坐标Y,绘制标准曲线,得回归方程、相关系数(r)和线性范围,见表3。

表3 各成分线性回归分析Tab. 3 Regression analysis of different components
2.5 精密度考察
精密吸取同一混合对照品溶液10 μL,按“2.1”项下色谱条件连续进样 6 次,记录各成分峰面积,计算原儿茶酸、原儿茶醛、牡荆苷峰面积的 RSD,分别为0.23%、0.28%、0.13%,表明仪器精密度良好。
2.6 稳定性考察
精密吸取同一供试品溶液(批号:20200528,昌江区乌蕨)10 μL,分别于0、2、6、10、16、20、24 h进样测定,记录各成分峰面积,计算原儿茶酸、原儿茶醛、牡荆苷峰面积的 RSD,分别为0.69%、0.61%、0.68%,表明供试品溶液在24 h内稳定性良好。
2.7 重复性考察
精密称取同一批样品(批号:20200528,昌江区乌蕨)共6份,按“2.3”项方法平行制备6份供试品溶液。精密吸取供试品溶液10 μL,按“2.1”项下色谱条件测定各成分含量,测得原儿茶酸、原儿茶醛、牡荆苷平均含量分别为0.335 7 mg/g、0.406 4 mg/g、2.443 4 mg/g ,RSD分别为1.15%、1.25%、1.06%,表明该法重复性良好。
2.8 加样回收率试验
取已知含量的乌蕨药材粉末(批号:20200528,昌江区乌蕨)6份,各约0.5 g,精密称定,分别精密加入含有原儿茶酸、原儿茶醛、牡荆苷的混合对照品溶液(原儿茶酸6.760 8 μg/mL、原儿茶醛9.493 1 μg/mL、牡荆苷39.28 μg/mL)25 mL,按“2.3”项下方法平行制备6份供试品溶液。精密吸取供试品溶液10 μL,按“2.1”项下色谱条件测定各成分含量,计算4种成分的加样回收率及RSD,见表4。
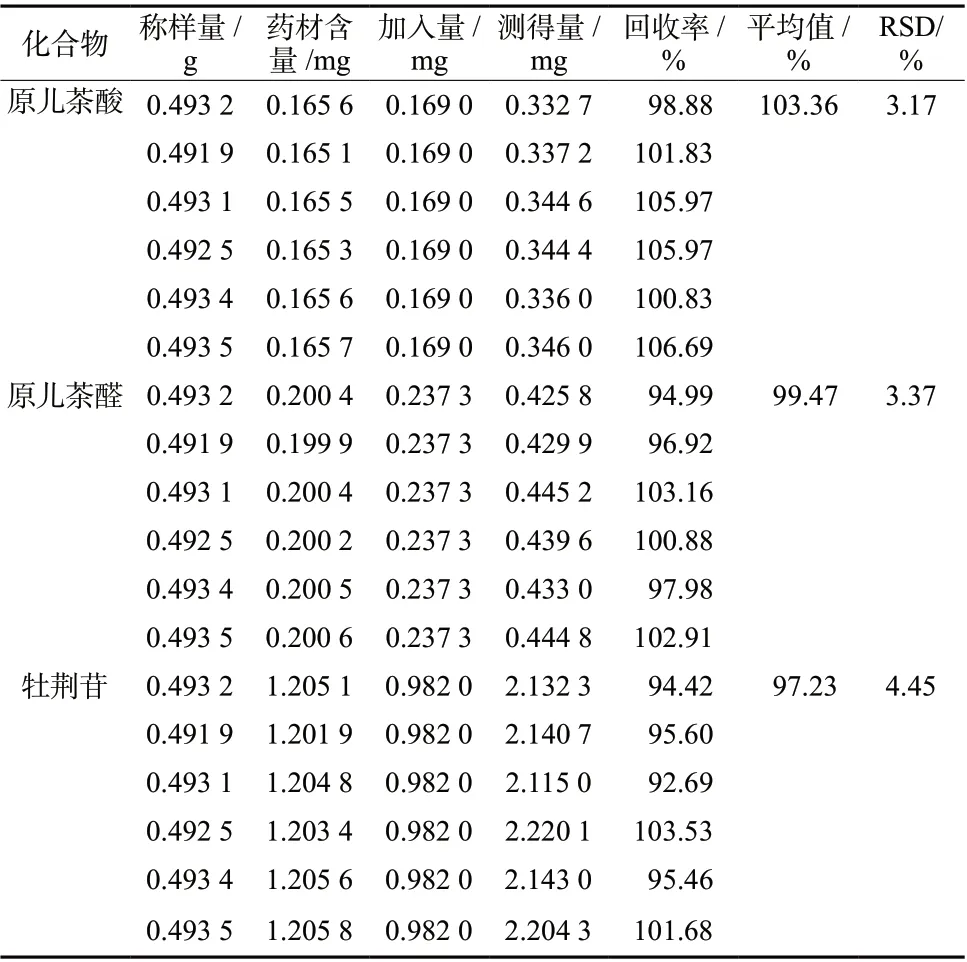
表4 回收率结果(n = 6)Tab. 4 Results of recovery rate( n = 6)
2.9 样品含量测定
取不同产地的乌蕨样品,按“2.3”项下方法制备供试品溶液,精密吸取供试品溶液10 μL,按“2.1”项下色谱条件测定各成分含量,计算3种成分的含量,见表5。从乌蕨样品中3种成分含量测定结果可知,牡荆苷的含量最高(0.598 9~2.443 4 mg/g),而原儿茶酸、原儿茶醛受生长环境、采收期等影响,含量差异较明显。以3种成分的含量及总含量为指标进行聚类分析,由图2可知,当样品含量分为4类时,渝水乌蕨(S2)单独聚为一类,总含量最高(3.608 5 mg/g);横峰乌蕨(S5)单独聚为一类,总含量最低(1.239 6 mg/g);而浮梁乌蕨(S3)和昌江乌蕨(S4)聚为一类;其它各为一类,说明其3种成分总含量较接近。
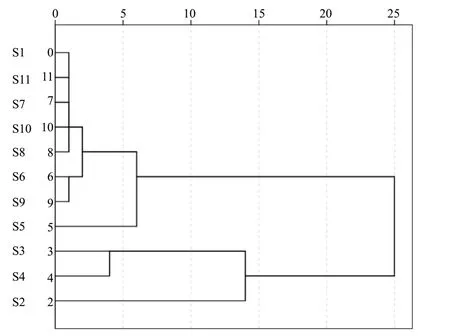
图2 11批乌蕨药材样品的聚类树状图Fig.2 Cluster dendrogram of 11 batches of Stenoloma chusanum samples

表5 不同产地乌蕨含量测定结果(mg/g,n = 2)Tab. 5 Results of content determination of different origin of Stenoloma chusanum (mg/g,n = 2)
3 讨论
3.1 流动相的选择
参考相关文献[14-15],本实验先后考察了甲醇-水、甲醇-0.1%磷酸水、甲醇-0.4%磷酸水、乙腈-水、乙腈-0.1%磷酸水、乙腈-0.4%磷酸水、乙腈-0.2%冰醋酸水等色谱条件对样品中待测成分的分离情况,结果显示有机相为乙腈时基线漂移较小且洗脱能力强;水相为0.4%磷酸水时,色谱峰分离效果好、峰形尖锐、对称性良好且理论塔板数高。综合分离情况、出峰时间、基线漂移程度、定量准确度等因素,最终确定以乙腈-0.4%磷酸水为流动相进行梯度洗脱。在该色谱条件下,样品分离度、重复性、稳定性良好,可用于乌蕨药材的质量控制。
3.2 供试品提取条件的优化
由于乌蕨中原儿茶酸、原儿茶醛、牡荆苷三种成分的极性差异较大,故考察了不同浓度甲醇和乙醇对待测成分提取的效果。除此之外,提取方式、提取时间、提取溶剂量三者对药材中有效成分的提取率影响也较大,因此本实验采用单因素考察法,以原儿茶酸、原儿茶醛、牡荆苷的含量为检测指标,对提取溶剂(70%甲醇、100%甲醇、65%乙醇、95%乙醇)、提取方式(回流、超声)、超声提取时间(30、60、90 min)、提取溶剂量(15、25、50 mL)分别进行了考察。结果表明,样品以70%甲醇超声提取效率高于其它溶剂与方法。在超声时间方面考察发现,待测成分提取率由大到小依次为:超声90 min>超声60 min>超声30 min,但三者无显著性差异,考虑到既节约能量又保证待测成分提取完全,最终选择样品超声60 min为最佳提取时间。
3.3 检测波长的选择
采用HPLC-DAD检测器分别对三种待测成分在190 ~ 400 nm波长处进行全扫描,结果表明,原儿茶酸最大吸收波长为260 nm;原儿茶醛最大吸收波长为270 nm,但在260 nm处也有较大吸收;而牡荆苷在340 nm处有最大吸收,在此波长检测下,样品中牡荆苷响应值高,图谱基线平稳且干扰峰少。考虑到待测成分间吸收波长差异较大,为确保3种成分均能得到较好的响应,因此采用双波长同时进行检测,以便能更准确地对3种物质含量进行测定。
4 结论
本研究建立了同时测定乌蕨中原儿茶酸、原儿茶醛、牡荆苷的HPLC双波长分析方法。结果显示,原儿茶酸等3个指标成分之间含量差异较大,原儿茶酸最大值可达最小值的3倍,原儿茶醛最大值可达最小值的4.5倍,牡荆苷最大值可达最小值的4倍,其中11批样品中又以牡荆苷含量最高,为0.598 9 ~2.443 4 mg/g。而导致各地乌蕨中活性成分含量差异大的原因,可能与植物自身遗传多样性、地理位置、生长环境、土壤、光照、气温、雨量等因素有关。该方法符合定量分析要求、操作简单、灵敏度高、重现性好,且在各成分适宜波长处分别测定其含量,可明显提高测定结果的准确度。本法可作为乌蕨药材含量测定方法,可为其质量控制和质量标准的建立提供参考。
