HTRA1相关常染色体显性遗传脑小血管病1例☆
2021-09-02陈莉刘欢邢晓娜陈晓虹
陈莉 刘欢 邢晓娜 陈晓虹
遗传性脑小血管病 (cerebral small vessel disease,CSVD)根据遗传形式可分为三类:显性型、X连锁型(Fabry病)和隐性型[1-5]。伴皮质下梗死和白质脑病的常染色体隐性遗传性脑动脉病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy,CARASIL)是由HTRA1的双等位基因纯合突变引起的,临床特征是青年期(20~45岁)发病,神经系统特点包括缺血性卒中(腔隙性脑梗塞、脑白质病变、微出血、脑萎缩)、认知障碍、情绪改变,非神经系统特点包括腰痛或脊椎发育障碍和40岁前的脱发症[6-7]。近年来,新发现的HTRA1基因杂合性突变可导致近5%的家族性CSVD,这些疾病呈现显性遗传模式,与CARASIL不同的是发病较晚、程度较轻且缺乏非神经系统特征[8-10]。本文报告1例以反复发作轻型卒中为主要表现的患者,无传统脑血管病危险因素,既往发育正常,通过分析患者神经影像及遗传学特点,总结报告该基因的临床表型。
1 临床资料
患者,男,49岁,近5年来反复发作性小卒中表现伴轻度颈腰部疼痛及间断头晕发作,自发病以来出现焦虑、抑郁情绪。既往无高血压病、糖尿病、冠心病等其他脑血管病危险因素。个人史:运动和智力发育正常,无烟酒等不良嗜好。家族史:否认父母近亲婚配史;患者母亲47岁患脑血管病于60岁死亡;患者母亲的三个兄弟姐妹患脑血管病死亡(见图 1)。
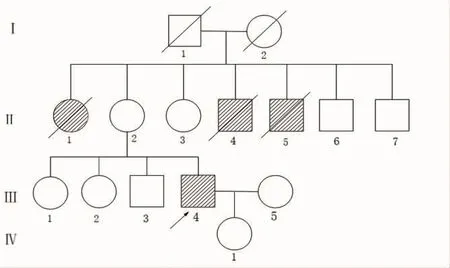
图1 患者家系图 箭头为先证者;条纹符号为脑血管病患者;方形为男性;圆形为女性;斜线为死者。
体格检查:血压127 mmHg/84 mmHg。无明显脱发表现。腰椎弯曲不受限,四肢关节无畸形和活动受限。心、肺、腹体检无异常。神经系统检查:双上肢及左下肢肌力5级,右下肢肌力4+级;左侧肌张力正常,右侧肌张力稍高。双上肢腱反射稍亢进,右>左;双下肢腱反射亢进。轮替动作可完成,右侧较左侧稍笨。Babinski’s征(左-,右+),余神经系统查体未见明显异常。
辅助检查:神经心理学测试示MMSE评分为30/30分,MOCA为27/30分(扣分项目为延迟回忆-2,语言-1),Fuld物品记忆测验 16分(>11分)、词语流畅性 35分(>28分)、Rey听觉词语学习测验35分 (>15分)、波斯顿命名30分(>24 分)、Stroop 色词测验 49 分(>39 分)、数字广度测试13分(>7分)均完成良好,无认知功能损害。眼底检查显示视乳头边界清,色淡红,动静脉之比为2:3。血脂、血糖、甲状腺功能等血液和相关免疫学检查均无异常。头MRI显示在FLAIR像可见在脑干、大脑皮层下、基底节多发腔隙性梗死灶 (见图2A、B);DWI示左侧脑室旁可见新发梗死灶 (见图2C);FLAIR像上双侧侧脑室前后角旁见广泛白质异常信号(见图2D);T2WI可见血管周围腔隙(见图2E);未见明显脑萎缩(见图2F);SWI像上呈多发点状微出血灶(见图2G、H);MRA示颅内大血管未见明显血管狭窄(见图2I)。脊柱MRI检查显示(见图3)颈椎、胸椎、腰椎的椎体及椎间盘退行性改变,各椎间盘不同程度突出,脊髓内无异常信号。双侧颈动脉、椎动脉超声及锁骨下动脉超声未见明显异常。心脏、肝、胆、胰、脾、甲状腺彩超检查未见异常。肌电图及诱发电位示双下肢胫、腓肠运动神经未见异常。

图2 患者头MRI 显示在FLAIR像可见在基底节、大脑皮层下多发腔隙性梗死灶 (A、B);DWI示左侧脑室旁可见新发梗死灶(C);FLAIR像上双侧侧脑室前后角旁见广泛白质异常信号(D);T2WI可见血管周围腔隙(E);未见明显脑萎缩(F);SWI像上呈多发点状微出血灶 (G、H);MRA示颅内大血管未见明显血管狭窄(I)。

图3 患者颈椎、胸椎和腰椎T2WI 多发椎体退行性变,椎间盘变性伴随椎间盘突出。
基因测序:对患者及其女儿、患者的2个姐姐和1个哥哥均进行了HTRA1基因直接测序,还对患者进行Notch3基因直接测序。结果:患者HTRA1基因第1号外显子存在C.267C>A杂合突变,该无义突变(又称截断突变)导致其编码蛋白第89位氨基酸(半胱氨酸)之后的392个氨基酸均无法翻译,从而使HTRA丝氨酸肽酶1缩短了392个氨基酸。其女儿HTRA1基因第1号外显子也存在同患者一致的C.267C>A杂合突变,患者的2个姐姐及1个哥哥未发现HTRAl基因突变。患者Notch3基因第13号内含子存在杂合突变C.2144+14T>C,而内含子突变是不会影响氨基酸编码的。
2 讨论
HTRA丝氨酸肽酶1(HTRA1)具有高度保守的类胰蛋白酶样丝氨酸蛋白酶结构域和至少1个C-末端PDZ结构域,它参与多种细胞过程,尤其是在转化生长因子-β信号通路中发挥重要的作用,通常被认为是血管生成和血管内稳态的调节因子[11]。HTRA1基因突变与多种疾病及病理生理有关如年龄相关性黄斑变性[12]、关节炎、常染色体隐性遗传性脑动脉病及动脉硬化伴皮质下梗死及白质脑病(cerebral autosomal recessive arteriopathy/arteriosclorosis withsubcortical infarcts anleukoencephalopathy,CARASIL)、Loeys-Dietz综合症和遗传性出血性毛细血管扩张等[13]。在CARASIL中HTRA1基因突变导致HTRA1酶活性丧失,不能抑制转化生长因子-β(transforming growth factor-β,TGF-β)家族信号,使纤连蛋白EDA结构域和多功能蛋白聚糖在患者脑小动脉内膜中积累使其增厚,进而引起管腔狭窄、动脉硬化和血管重塑,导致缺血性小血管疾病,TGF-β也调控头发毛囊的发育和骨骼形成,导致了脊椎病和脱发的临床表现[14]。
在一些CARASIL的病例报道中发现先证者的HTRA1基因杂合子突变的亲属都具有不同程度的脑白质病变[15-17]、轻度脱发[17]及脑血管病等临床表现[15-17]。此外在一些HTRA1基因杂合突变相关性CSVD的病例报告[18-22]中也发现患者有类似CARASIL的临床表现,尤其是头MRI上表现出的多发脑白质及微出血改变[14],随访研究中发现此类患者在神经系统检查、头MRI和神经心理检查中具有一定的临床和影像学的稳定性[21]。在一些对大量不明原因的家族性CSVD进行的研究[8-10]中发现有些HTRA1基因突变表现为显性突变,杂合子携带者在临床上受到影响,其频率预计远高于隐性型突变,这些突变占家族性脑小血管病的近5%。还发现这些HTRA1基因杂合突变体发病年龄迟于、临床表现不同、致病等级不同且远低于具有双等位突变的后代,这些表现为显性遗传模式,与CARASIL不同之处在于晚发和有时缺乏非神经学特征,即CARASIL杂合患者的所谓“弱”表型。
本病例是新型杂合HTRA1基因突变的携带者,为显性遗传模式,先证者的兄弟姐妹都无该基因突变,遗憾的是由于患者父母及母亲的两个兄弟已经死亡,缺少检测信息,无法判断遗传共分离。先证者女儿存在与其相同的HTRA1基因杂合突变,可能是从先证者那里继承而来,也有可能是突变而来。先证者的杂合突变体为新发现的突变位点(在MAF数据库中未收录),根据美国医学遗产学和基因组学与分子病理学协会指定的解读指南进行的分级提示其具有致病作用,符合三个证据PM、PM2和PVS1。先证者临床表现为反复小卒中伴轻微腰痛,类似CARASIL的临床表现但程度较轻且发病较晚,头MRI具有典型的脑小血管病神经影像学表现包括广泛脑白质病变、多发腔隙性脑梗死、脑微出血、周围血管间隙,颈、胸及腰椎多发椎间盘突出,因此本研究支持这种HTRA1基因杂合突变相关CSVD表现为典型CARASIL较轻和迟发表型的理论。
杂合HTRA1相关CSVD的分子机制尚不完全清楚,学者们对家族性CSVD进行基因分析后[8-10]认为不同位点、部位、突变类型都会导致HTRA1酶活性丧失或降低,但降低程度不同,失活机制也不同。对HTRA1酶活性降低或丧失机制的提出了一些猜测[8]认为一些错义突变可能是占优势的负突变,突变的等位基因是功能缺失突变体,扰了剩余野生型等位基因的正常功能,导致酶活性进一步下降;也可能其中一些错义突变是半显性突变;或者突变导致过早终止密码子将导致突变的mRNA的衰变,突变等位基因编码的蛋白质缺乏合成,从而导致单倍功能不全,其本身不足以引起CSVD,这可以解释有些杂合携带者在临床上不受影响。有研究统计35个杂合突变发现氨基酸最接近250-300的氨基酸受到的影响最大,其次是氨基酸150-200,突变位点区域集中的趋势可能与这些区域中负责HTRA1相关常染色体显性CSVD发病机理的关键位点的浓度有关[22]。本病例HTRA1基因的该无义突变导致其编码的蛋白第89位氨基酸之后的392个氨基酸均无法翻译,因此其发病机制可能为半显性突变导致HTRA1酶活性降低,也可能与其无法翻译的氨基酸恰巧位于负责HTRA1相关常染色体显性CSVD发病机理的关键位点有关。
本病例扩大了与常染色体显性小血管病临床表型有关的HTRA1基因中新杂合突变的范围,为携带HTRA1基因杂合突变的CSVD患者的临床模式提供了一定的资料。杂合HTRA1基因突变可能是CSVD的病因,因此建议对所有不明原因的CSVD患者都应进行HTRA1基因的筛选。
