肌萎缩侧索硬化SOD1基因多态性与临床表型分析☆
2021-09-02吴瑞鹏冯斌任占宏谷成高福林张毅
吴瑞鹏 冯斌 任占宏 谷成高福林 张毅
肌萎缩侧索硬化(amyotrophic lateral sclerosis muscular atrophy,ALS) 分为家族性 ALS (familial amyotrophiclateral sclerosis,FALS)和散发性 ALS(sporadic amyotrophic lateral sclerosis,SALS)。关于ALS的基因组学特征,现阶段研究较明确的有铜/锌超氧化物歧化酶1(Cu/Zn superoxide dismutase,SOD1)、 反式激活反应-DNA结合蛋白 (TARDBP)基因、C9orf72基因、肉瘤熔合(FUS)基因,占到家族性 ALS的20%,占到散发型ALS的1%[1-2]。与SOD1基因相关的亚型称为ALS1亚型,目前SOD1基因已经报告了200余种基因突变类型[3],中国ALS人群中的研究共报告了SOD1基因突变26种[4-8],国内报告较多的SOD1基因突变H46R 、V47A、G37R、Cys111Tyr和 Gly147Asp,多集中于华北地区及华东地区[9]。甘肃为亚高原,多民族杂居,ALS基因组学有其特征性表现,本研究收集甘肃地区FALS患者家系进行SOD1基因突变检测,分析SOD1突变患者的临床特点及突变与临床表型关系。
1 对象与方法
1.1 研究对象纳入2018年1月至2020年6月收治的18例FALS家系的先证者,男7例,女11例,平均年龄(48.3±12.6)岁。所有病例由至少两位以上神经内科专科医生独立做出诊断,诊断标准依据EI Escorial修订诊断标准[10]。排除标准:①有肿瘤疾病史;②有严重心脑血管疾病;③精神异常及外伤病史;④有其他家族遗传病史。所有研究对象签署知情同意书,本研究得到我院伦理委员会批准。
1.2 方法
1.2.1 外周血DNA提取 抽取先证者外周血2 mL,酸性柠檬酸葡萄糖溶液(ACD)抗凝后,用 Qiagen试剂盒进行DNA提取,无水乙醇洗涤两次,-80℃保存。
1.2.2 SOD1基因PCR扩增引物及反应条件

表1 SOD1基因PCR扩增引物及反应条件
1.2.3 DNA序列分析 PCR产物Qiaquick柱纯化,采用美国ABI公司生产的3730 DNA自动测序仪进行 DNA序列分析,测序结果与美国国立生物技术信息中心(NCBI)数据库中心公布的人类基因组SOD1 cDNA序列进行比对。
2 结果
18例先证者中,3例检测出SOD1阳性,均为杂合突变,其中 2例 p.Gly73Ser突变,1例p.G42D突变,其余15例未检测出SOD1基因突变,SOD1基因阳性突变检出率为 16.67%(3/18例)。
2.1 先证者SOD1基因突变的家系分析p.Gly73Ser突变FALS家系临床资料如下。A(图 1A):先证者(Ⅲ6)为 46岁女性,以缓慢进展的右上肢无力为首发症状,患者于2017年8月发现右手拇指和食指捏握无力,数月后出现部位不固定的肌束颤动,无疼痛及其他不适感觉,2018年3月,发现右上肢肌肉萎缩,以大鱼际肌及三角肌萎缩明显。期间就诊多家医院,于2019年7月在我院就诊,患者出现左手力弱,伴行走时右足活动不灵活,伴有活动后气短,查体发现右手及上臂肌肉无力、萎缩,锥体束症状不明显。肌电图提示脊髓颈段、胸段神经源性损害。先证者两子目前无发病,先证者叔叔(Ⅱ2)于57岁时因呼吸衰竭过世,于45岁时开始出现腿部无力,病情缓慢进展,9年后开始使用轮椅,期间就诊多家医院诊断ALS,发病10余年后因呼吸衰竭行气管切开辅助呼吸机通气,半年后过世。先证者父亲(Ⅱ4)于63岁时死于结肠癌并脑转移,当时结肠癌确诊5年,已行手术切除,期间无运动神经元病的表现。先证者母亲(Ⅱ5)现已 76岁,目前身体健康。B(图 1B)先证者(Ⅲ3)为50岁女性,4年前发现双手持物困难,左侧重,1年多后出现走路不稳,上下楼梯困难,后逐渐出现左上肢鱼际肌、前臂肉跳,无球部症状,肌电图提示广泛神经源性损害(脊髓颈胸腰段),全脊髓MR无异常。先证者母亲(Ⅱ2)病史7年,后因继发肺部感染过世。先证者叔叔47岁死于车祸伤。

图1 p.Gly73Ser突变的FALS家系图(A、B图)
p.42GD突变FALS家系临床资料如下。先证者 (Ⅳ1)为45岁女性,病史8年,她在37岁时出现走路摇晃、不稳,之后的4年多病情缓慢进展,42岁时出现双腿僵硬感,自觉右腿比左腿严重,43岁时需手杖行走,44岁时行走需要轮椅,同时出现上肢肌无力伴随吞咽困难,半年后出现声音嘶哑、呼吸急促,伴弓形足及肌束震颤,全脊髓MR、血液、脑脊液检查均无异常。肌电图显示神经源性改变。先证者母亲(Ⅲ1)健康,父亲(Ⅲ2)因30多年的单侧下肢肌萎缩,一直未明确诊断,其基因型为p.G42D杂合突变。家族史中的一位叔叔(Ⅲ3)于70岁时死于散发ALS。其弟(Ⅳ3)发育迟缓,其小姨(Ⅲ10)和表弟(Ⅳ4)诊断为遗传性痉挛性截瘫(图2)。
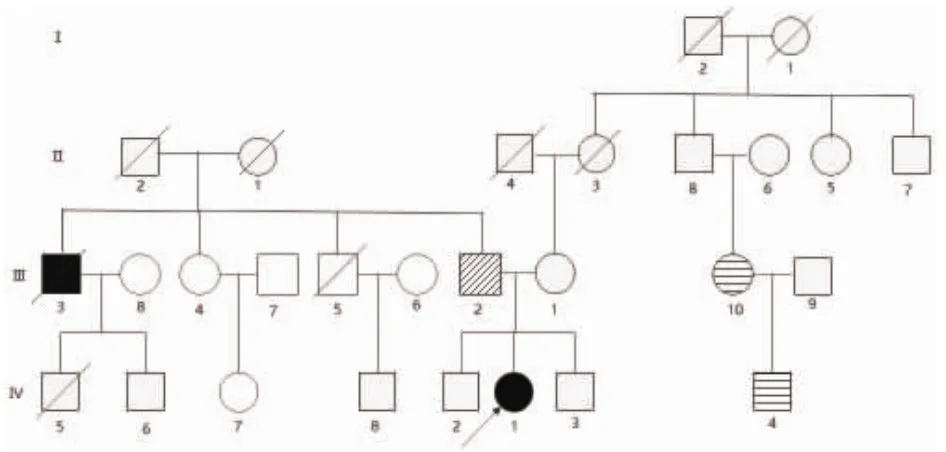
图2 p.G42D突变的FALS家系图
2.2 SOD1基因突变结果
图3 对先证者应用NGS+PCR-Sanger测序法对SOD1基因5个外显子和侧翼序列的突变筛查发现,突变位于3号外显子的杂合突变,cDNA在217位碱基由G突变为A,使第73位氨基酸由甘氨酸变为丝氨酸,如图3。

图3 SOD1基因外显子3的 p.Gly73Ser(c.217G>A)杂合变异
图4 对先证者应用NGS+Sanger测序法对SOD1基因5个外显子和侧翼序列的突变筛查发现,突变位于2号外显子的杂合突变,cDNA在125位碱基由G突变为A,使第42位氨基酸由甘氨酸变为天冬氨酸,如图4:p.G42D(c.125G>A)。
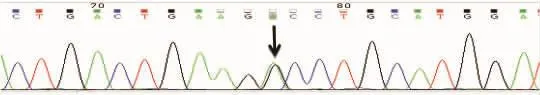
图4 SOD1基因外显子2的 p.G42D(c.125G>A)杂合变异
3 讨论
ALS是一种累及上下运动神经元的慢性退行性疾病,临床表现肌肉无力萎缩,椎体束征等,疾病后期出现严重的肌肉萎缩、延髓麻痹,最终多因呼吸衰竭或继发肺部感染死亡。目前病因、病理机制仍不明确,针对病因的研究多集中在兴奋性氨基酸毒性、自由基蓄积、遗传机制等,遗传机制目前得到广泛认可。在ALS中约有10%FALS,大多数FALS为常染色体显性遗传,目前已有30多个基因与FALS相关,SOD1的突变率国外报告仅有20%[11],本研究中18例先证者中,SOD1阳性概率仅为16.67%,与陈嬿等[6]报告的23.06%、张华纲等[7]报告的23.3%较低,提示SOD1基因在甘肃地区FASL患者中阳性率偏低。本研究中共收集FALS患者18例,汉族占15例,回族占1例,东乡族 1例,藏族1例,仍以汉族为主要发病群体,少数民族占16.67%,汉族仍是FALS发病的主要群体。SOD1基因蛋白是一种自由基清除酶,在神经组织、红细胞及肝脏中呈现高表达,具有抗氧化作用。目前认为SOD1基因突变导致蛋白毒性功能的获得是SOD1基因的致病机制[8],种族差异对SOD基因突变的影响目前国内尚无文献报告。我们猜测甘肃地区虽为亚高原地区,多民族杂居,除饮食结构有差异外,环境、物理因素、生活习惯均被汉化,因此种族差异对基因影响较小。
本研究中p.Gly73Ser突变的临床表型:先证者以肢体无力起病,以下运动神经元损害为主,病情进展缓慢,最终导致呼吸衰竭,病史达10年之久。ORRELL等[12]在一个ALS家系中检出 SOD1基因 Gly72Ser(同 p.Gly73Ser)杂合突变,红细胞检测结果显示SOD1酶活性降低45%,SHAW等[13]在1例ALS患者中检出SOD1基因Gly72Ser突变,此患者以下肢痉挛无力起病,最终呼吸衰竭,病程2年。这一类型临床表现以下肢起病,上运动神经元损害不明显,易误诊为周围神经病变,且目前国内尚无此突变基因的报告。p.G42D突变发病年龄较小,30多岁起病,病情进展缓慢甚至处于相对静止状态,以下肢起病为主,病史长达30年。由于先证者父系和母系可能存在交叉影响,导致先证者弟弟幼年时期出现智力低下,具体基因型尚不明。
目前在人类基因库中报告了200多个SOD1基因突变,其临床表现多样化,从急性进展性到缓慢演变,其进展方式也各有不同[14]。本研究对甘肃地区18个FALS家系筛查SOD1基因突变,检出Gly72Ser和p.G42D两种突变,这一结果扩大了中国FALS患者的SOD1基因突变谱。同时通过对SOD1基因突变与临床表型关系的探讨,为更准确诊断FALS及判断预后提供依据,也为ALS患者及其亲属提供基因学的遗传咨询。
