肾透明细胞癌病理分级相关基因的WGCNA筛选
2021-08-03宋章兴崔应东李时军谭威向圣坎向奎
宋章兴 崔应东 李时军 谭威 向圣坎 向奎
肾细胞癌(renal cell carcinoma, RCC)是成人肾肿瘤最常见的恶性肿瘤,占所有成人肾肿瘤的90%以上,大约1/3的RCC患者诊断明确时已经有远处转移,尽管目前的检测和治疗取得了很大的进展,但RCC的总体生存率仍然很低[1]。肾透明细胞癌(clear cell renal cell carcinoma, ccRCC)占RCC的75%~85%,是其最常见的亚型,ccRCC通常对放化疗有抵抗性,靶向疗法因其靶点特异性和低毒性而被采用,可能成为非手术治疗的最佳选择[2-3]。目前发现的可以预测ccRCC治疗效果和临床预后的生物标志物,包括VHL、VEGF、CAIX和HIF1α/2α突变[4]。ccRCC的致癌机制可能涉及多因子、多基因协同的复杂过程,包括多种致癌基因或肿瘤抑制因子失控等[5]。因此,迫切需要鉴定出可以预测ccRCC患者疾病阶段和临床结果的新型分子生物标志物,从而有助于了解其发病机制并提供个性化治疗。加权基因共表达网络分析(weighted gene co-expression network analysis, WGCNA)算法可以构建自由尺度的基因共表达网络,以探索不同基因集之间或基因集与临床特征之间的关系[6]。本研究中,采用WGCNA分析ccRCC患者临床特征和基因芯片数据,构建共表达网络筛选与ccRCC病理分级相关的核心基因,以期为ccRCC的诊断和预后提供新的生物标志物或治疗靶点。
对象与方法
一、基因表达数据集
从基因综合表达(Gene Expression Omnibus, GEO)数据库下载芯片数据集,基于芯片平台为GPL10558的GSE40435(2013 捷克)作为训练集进行WGCNA的构建,该数据集包括101例癌组织及配对的癌旁组织,临床特征包括年龄、性别、病理分级。芯片基于芯片平台为GPL570的数据集GSE73731(2017 美国)作为测试集,该数据集包括265例癌组织,临床特征包括年龄、病理分期、病理分级。基于癌症和肿瘤基因图谱(The Cancer Genome Atlas, TCGA)数据库可视化分析工具GEPIA中的623例样本,其中523例ccRCC及100例癌旁组织数据作为独立的第二个测试集[7]。
二、数据整理和基本信息
本研究中的GEO数据集采用R软件Affy包读取原始CEL文件,采用RMA算法对数据进行背景过滤、归一化、标准化处理后得到表达矩阵,再进行后续处理;GSE40435训练集筛除临床数据中为空值的样本,同时保证GSE73731测试集病理分级的数据无空值。GSE40435训练集及GSE73731测试集进行标准化等处理,提取相关重要临床数据。GSE40435训练集包括101例癌组织及配对的癌旁组织,101例癌组织样本的临床信息:年龄为(64.11±9.22)岁,其中男59例、女52例;病理分级为1级22例,2级47例,3级24例,4级8例。GSE73731测试集包括265例癌组织,其中256例癌组织包含完整数据的病理分级:1级22例,2级90例,3级95例,4级49例。
三、统计学分析
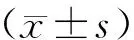
2.差异基因分析:训练集GSE40435的表达矩阵,通过计算样本之间的Pearson相关系数进行聚类分析,去除离群样本后,采用R软件limma包分析ccRCC与癌旁组织获得差异表达基因(differentially expressed genes, DEGs),筛选条件为校正后的P值,即错误发生率(false discovery rates, FDR)<0.05和对数化表达倍数(︱log2FC︱)≥0.585。
3.WGCNA的构建:使用R软件包WGCNA包,通过一步法构建DEGs的加权基因共表达网络。通过计算基因间的Pearson相关系数确定最佳的软阈值β,从而使网络更逼近无尺度网络。然后通过计算把邻接矩阵转化为拓扑重叠矩阵(TOM),并计算基因间的相异度dissTOM=1-TOM,进行层次聚类。最后使用动态剪切的方法合并相似模块。
4.核心基因的筛选:模块与临床特征关联后,通过计算模块显著性(module-significance, MS)确定显著性模块,MS值越高,提示该模块越重要。以显著性模块中模块身份(module membership, MM)>0.8及基因显著性(gene significance, GS)>0.2为筛选条件筛选基因。构建显著性模块内基因的蛋白互作网络(protein-protein interaction, PPI),显著性模块内的基因导入STRING数据库构建PPI网络,同时采用Cyctoscape软件进行可视化,设置基因间的连接度>10为筛选基因的条件。网格网络筛选基因的共有基因最终确定为核心基因。
四、核心基因的验证
使用验证数据集GSE73731的表达矩阵及临床特征进行线性回归分析,验证核心基因的表达水平与ccRCC之间的关系。使用TCGA可视化分析工具GEPIA中的623例样本(包含523例ccRCC及100例癌旁组织)数据验证核心基因在ccRCC中的表达水平、病理分期及预后情况。进一步采用人类蛋白质表达图谱,利用免疫组化分析核心基因在正常肾组织和癌组织的蛋白水平。
五、基因富集分析(gene set enrichment analysis, GSEA)
以GSEA官网MSigDB数据库中的h.all.v6.2.symbols.gmt [Hallmarks] 数据集作为功能基因集,置换次数为1 000次,设定P<0.05、FDR<0.25的基因集作为显著富集基因集,分析核心基因可能相关的生物学功能。
结 果
一、DEGs的筛选
GSE40435训练集通过去离群后发现GSM994069、GSM994041、GSM993996、GSM994065明显偏离,踢除离群样本后,GSE40435训练集最终纳入194例(癌组织97例,癌旁组织97例)样本进行差异基因分析,见图1。通过设置的筛选条件,得到2 546个DEGs,其中1 207个基因上调,1 339个基因下调。

图1 在GSE40435训练集的去离群
二、加权基因共表达网络构建及显著性模块确定
对GSE40435训练集中2 546个DEGs进行构建共表达网络。导入97例癌组织临床信息,与2 546个DEGs表达谱矩阵进行聚类分析,以用于后续分析,见图2A。选择β=9(R2=0.91)最为软阈值(图2B、2C)。确定软阈值后,采用动态剪切法,获得9个模块,见图3A。进一步分析各模块的特征向量基因(module eigengene, ME)及MS,发现红色模块包括86个基因,与ccRCC中的病理分级相关性最高(R2=0.56),红色模块的MS也是最高的,确定为显著性模块,见图3B、3C。

A:去离群后肿瘤样本及对应临床特征聚类树,图中颜色深浅代表数字对应的临床信息分类取值大小;B:不同软阈值对应的R2,确定β=9为软阈值;C:不同软阈值对应的基因邻接系数的均值图2 WGCNA样本与表型聚类及软阈值的确定
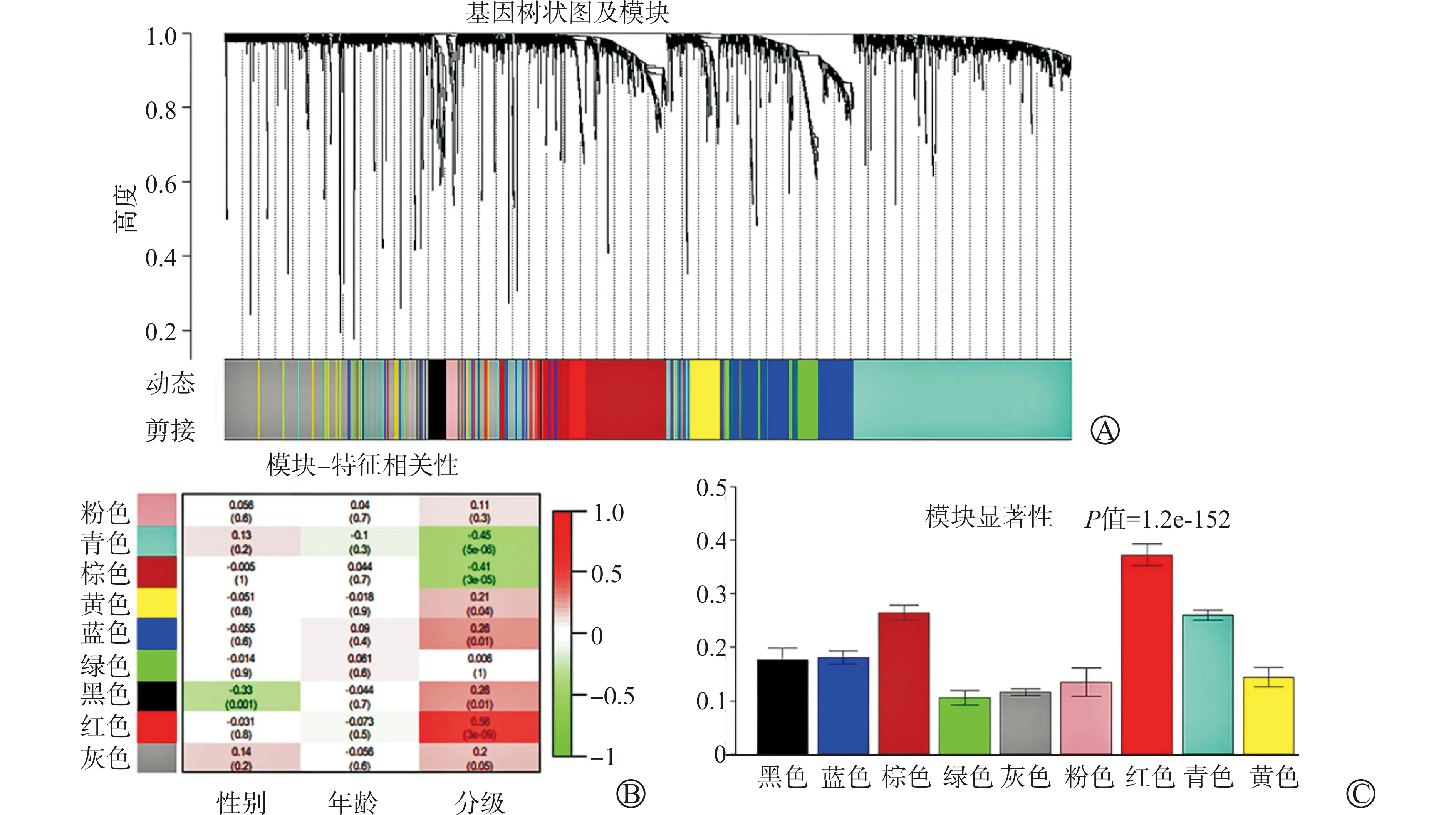
A:模块合并后WGCNA聚类树;B:基因模块ME与临床特征的相关性,颜色越深表示相关性越大,相关系数为正表示正相关,为负表示负相关;C:各模块MS分布柱状图图3 WGCNA网络构建及模块识别
三、核心基因的确定
在红色模块中,通过︱MM︱>0.8和︱GS︱>0.2筛选得到17个备选核心基因。同时把该模块中的86个基因导入STRING数据库,进行PPI网络构建,以连接度>10筛选得到14个备选核心基因,取二者的交集获得TOP2A、PTTG1、PRC1、UHRF1这4个基因为最终的核心基因,见表1。

表1 筛选关键基因
四、核心基因的验证
使用GSE73731测试集及GEPIA数据库分析各核心基因与ccRCC病理分级及病理分期的关系,结果显示各核心基因与ccRCC病理分级及分期呈正相关,验证了训练集的分析,见图4。使用GEPIA数据集中的623例样本(其中523例ccRCC及100例癌旁组织)数据验证核心基因在ccRCC的表达水平及预后情况,结果显示各核心基因在癌组织中表达显著升高,同时各核心基因高表达则OS及无病生存期(disease free survival, DFS)均较差,见图5。人类蛋白质表达图谱分析结果显示TOP2A与UHRF1在癌组织中呈中强度表达,PTTG1与PRC1在癌组织中呈高强度表达,见图6。

A:GSE73731测试集及GEPIA数据库分析核心基因与ccRCC病理分级的关系;B:GEPIA数据库分析核心基因与ccRCC分期的表达水平及预后情况图4 核心基因与ccRCC病理分级及分期的关系
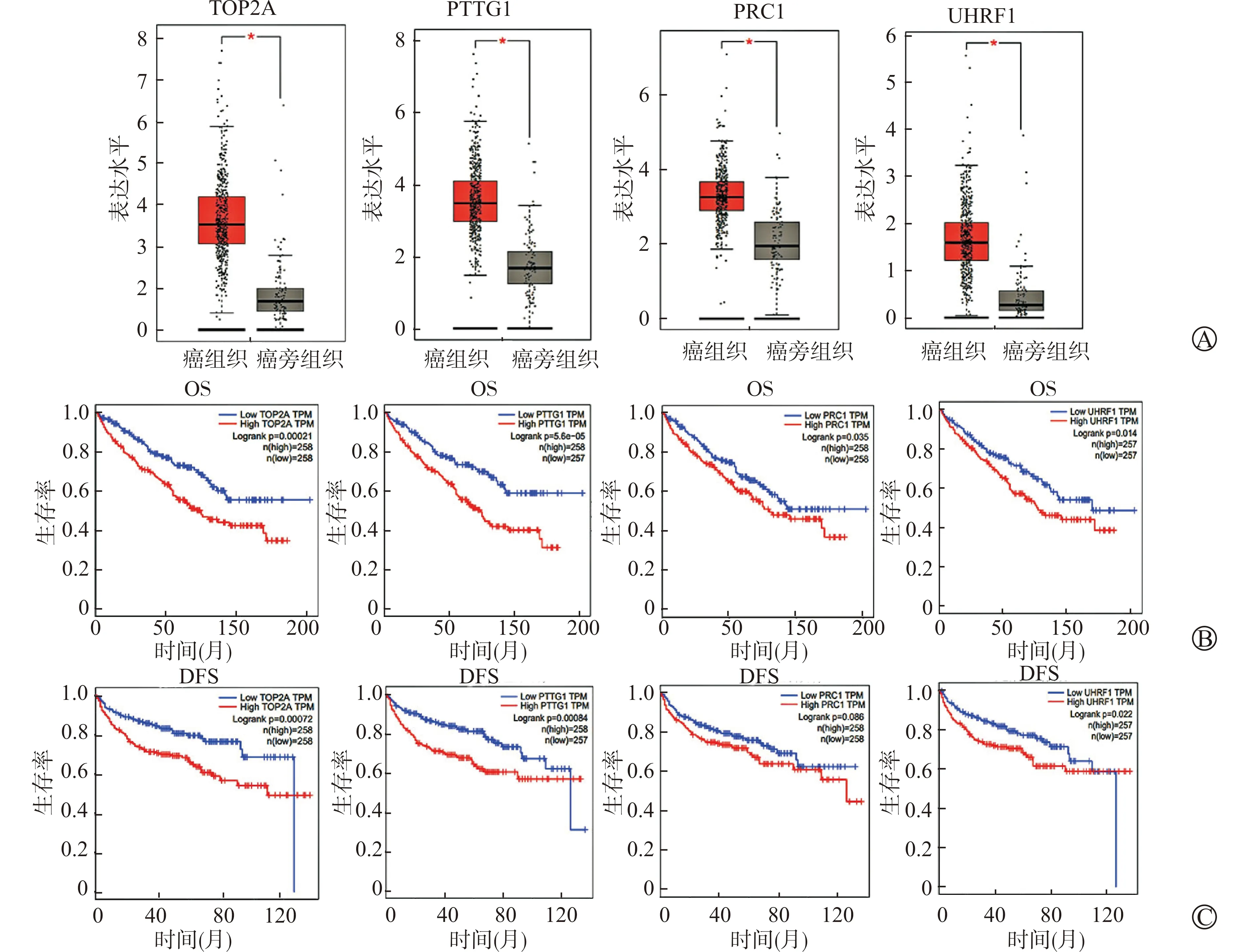
A:GEPIA数据库分析核心基因在ccRCC的表达水平;B:GEPIA数据库分析核心基因在ccRCC的OS;C:GEPIA数据库分析核心基因在ccRCC的DFS图5 GEPIA数据集验证核心基因在ccRCC的表达水平及预后情况

图6 人类蛋白质表达图谱分析各核心基因在肾组织和癌组织中的蛋白水平
五、核心基因的GSEA分析
核心基因以GSEA官网MSigDB数据库中的h.all.v6.2.symbols.gmt [Hallmarks] 数据集进行分析,结果提示,核心基因主要富集了E2F、有丝分裂纺锤体信号通路及G2M检查点等生物学过程,提示核心基因可能通过以上生物学过程促进肿瘤细胞增殖,影响患者临床及预后,见表2。

表2 核心基因的GSEA分析
讨 论
ccRCC预后差,给患者带来巨大的负担。在精确的医学时代,迫切需要更好的生物标志物用于癌症的预后和进展,从而可以提高患者早期诊断及治疗的决策能力。WGCNA已被广泛应用于寻找与不同癌症类型的临床特征相关的核心基因[8-9]。 WGCNA相较于单独的差异基因分析,可以对基因之间的高度互连网络进行整体分析,同时识别基因或模块簇与样本外部特征之间的关系[10]。因此在本研究中,我们使用了该方法进行综合分析来筛选ccRCC进展和预后相关的生物标志物。
本研究利用WGCNA识别出红色模块(包括86个基因)与ccRCC的病理分级相关性最高(R2=0.56),确定红色模块为显著性模块,进一步结合PPI网络,挖掘出TOP2A、PTTG1、PRC1、UHRF1这4个核心基因。GSE73731测试集及GEPIA数据库验证了核心基因与ccRCC的病理分级及分期呈正相关。GEPIA数据库验证了核心基因在ccRCC中高表达,同时初步探讨了核心基因对ccRCC预后的影响,发现高表达核心基因组的ccRCC预后差。TOP2A称作DNA拓扑异构酶Ⅱα,该基因编码为DNA拓扑异构酶,是一种在转录过程中控制和改变DNA拓扑状态的酶。这种核酶参与了诸如染色体浓缩、染色单体分离和DNA转录和复制过程。Liu等[11]报道MDM4和TOP2A相互结合,并在翻译后水平相互上调,导致TOP2A蛋白稳定,抑制p53,促进肿瘤细胞增殖,结果揭示了MDM4和TOP2A的新功能以及它们在肿瘤发生中的相互作用,提示抑制MDM4-TOP2A的相互作用可能是一种同时特异性地针对TOP2A和MDM4癌症治疗的新策略。Zhang等[12]研究发现LncRNA SNHG3通过上调TOP2A促进ccRCC的增殖和迁移,初步表明了TOP2A在ccRCC中可能的作用。PTTG1称作垂体肿瘤转化基因1,该基因编码的蛋白是酵母securin蛋白的同源物,可阻止separins促进姐妹染色单体分离,作为一种细胞周期后期促进复合物(APC)底物,可与separin结合直至激活APC。PTTG1产物具有体外转化活性和体内致瘤活性,并且该基因在各种肿瘤中高表达[13]。Wei等[14]通过转移组及非转移组ccRCC的差异基因分析,发现PTTG1在ccRCC组织中表达上调,且与预后相关。Hu等[15]报道MicroRNA-329介导的PTTG1下调使丝裂原激活蛋白激酶(MAPK)信号通路失活,抑制了胆管癌细胞的增殖和肿瘤生长,为胆管细胞癌的靶向治疗提供依据。PRC1称作细胞分裂蛋白调节剂1,该基因编码参与胞质分裂的蛋白质,在有丝分裂的S和G2/ M阶段以高水平存在,但是当细胞退出有丝分裂并进入G1阶段时,其水平急剧下降。Liao等[16]报道,PRC1基因沉默可能通过抑制Wnt/β-catenin途径使细胞增殖和侵袭能力降低,G0/G1期延长,S期和G2/M期缩短,从而抑制视网膜母细胞瘤细胞的增殖和血管生成。Wu等[17]发现PRC1通过P53/PRC1/EGFR信号通路调控口腔鳞癌细胞增殖和细胞周期参与口腔鳞癌的发生、发展,为口腔鳞癌的靶向治疗提供了依据。UHRF1称作靶向泛素样含PHD和环指域1,该基因编码环指域E3型泛素连接酶亚家族的成员,与特定的DNA序列结合,并招募组蛋白去乙酰化酶来调节基因表达,在调控细胞周期及p53 依赖性 DNA 损伤检查点中发挥作用。Lu等[18]研究提示UHRF1通过诱导细胞增殖促进黑色素瘤的进展,并与黑色素瘤患者的不良预后相关。Ma等[19]发现UHRF1调控ccRCC p53泛素化和p53依赖性细胞凋亡,促进ccRCC的进展。以上相关研究支持挖掘的4个核心基因可能在ccRCC进展及预后中的作用,TOP2A、PTTG1、UHRF1在ccRCC的相关研究主要也是基于生物信息学分析,表明对ccRCC的进展存在重要作用,但具体的机制仍不十分明了,PRC1对ccRCC的作用尚未见报道。本研究进一步对这4个核心基因进行人类蛋白质表达图谱分析,提示TOP2A、PTTG1、PRC1、UHRF1在癌组织的蛋白水平较肾组织高表达,同时GSEA分析其机制可能通过E2F、有丝分裂纺锤体信号通路及G2M检查点参与ccRCC的生物学过程。
本研究通过WGCNA及PPI网络的方法识别并验证与ccRCC进展和预后相关的4个核心基因(TOP2A、PTTG1、PRC1、UHRF1),其可能作为ccRCC的候选生物标志物。
