基于GBS测序的全基因组SNP揭示贵州地方茶组植物资源的亲缘关系
2021-08-02郭灿皮发娟吴昌敏高秀兵乔大河周媛
郭灿 皮发娟 吴昌敏 高秀兵 乔大河 周媛
摘要:【目的】分析貴州地方茶组植物资源的亲缘关系,为明确贵州地方茶组植物资源的遗传关系及其保护、利用提供科学依据。【方法】以从贵州省内不同区域收集的41份茶组植物资源及贵州省茶叶研究所茶树种质资源圃保存的18份省内外育成茶树品种为材料,利用基于GBS(Genotyping by sequencing)的简化基因组测序技术对其基因组SNP位点进行检测,基于获得的高质量SNP位点对这些材料进行遗传特征分析。【结果】从59份茶组植物材料获得45.84 Gb高质量序列(Clean reads)数据,平均每个材料为795.6 Mb,约占改良版茶树基因组大小(2.93 Gb)的26.5%,平均比对率为72.62%,经过滤后得到248772个高质量SNP位点,其中83.98%的高质量SNP位点分布在基因间区,16.02%分布于基因区;有22614个SNP位点分布在内含子,15038个SNP位点分布在外显子区,2203个SNP位点分布在非翻译区(UTR)。59份茶组植物材料的观察杂合度(Ho)为0.016~0.081,期望杂合度(He)为0.006~0.064,F为-0.331~0.737。主成分分析结果、系统发育进化树构建情况及遗传结构分析结果均显示59份茶组植物材料可分为3个类群,其中全部茶种(Camellia sinensis)材料归在一个类群、疏齿茶(C. remotiserrata)和大厂茶(C. tachangensis)归在一个类群、9份突肋茶(C. costata)单独归在一个类群,但疏齿茶与大厂茶及两个区域的大厂茶均处于独立的亚类群,此外茶种中的阿萨姆变种(C. sinensis var. assamica,CSA)和中国变种(C. sinensis var. sinensis,CSS)也处于不同的进化分支;突肋茶与疏齿茶和大厂茶的亲缘关系较其与茶种的亲缘关系更近。茶种、疏齿茶、大厂茶和突肋茶4类茶组植物存在互相融合的遗传背景。根据地理来源和种质类型可将59份茶组植物材料分为11个种群,不同种群间具有较低的基因流,但种群内部具有较高的基因流。【结论】不同茶组植物在基因组水平上存在明显差异,经典形态学分类上合二为一的大厂茶和疏齿茶在遗传结构上也存在差异,即分为2个变种更合适;茶组植物种内亲缘关系与其地理来源直接相关,在育种实践中应尽量避免相同地理来源材料间的杂交应用。
关键词: 茶组;茶树;SNP;基因组;系统进化;遗传结构;基因流
中图分类号: S571.102.4 文献标志码: A 文章编号:2095-1191(2021)03-0660-11
Genome-wide SNP developed by genotyping-by-sequencing revealed the phylogenetic relationship of Sect. Thea (L.) Dyer resources in Guizhou
GUO Can1, PI Fa-juan2, WU Chang-min3, GAO Xiu-bing1*, QIAO Da-he1, ZHOU Yuan4
(1Institute of Tea, Guizhou Academy of Agricultural Sciences, Guiyang 550006, China; 2Bureau of Agriculture and Rural Affairs of Meitan, Meitan, Guizhou 563100, China; 3 Tea Industry Technical Service Center of Liping,
Liping, Guizhou 557300, China; 4Fengle Tea Co., Ltd., of Sandu, Sandu, Guizhou 558100, China)
Abstract:【Objective】To clarify the genetic relationship of local Sect. Thea(L.) Dyer resources in Guizhou,and to provide scientific basis for their protection and utilization. 【Method】A total of 41 Sect. Thea(L.) Dyer resources collected from different regions of Guizhou and 18 tea plant varieties bred from Guizhou and other provinces that kept by Institute of Tea,Guizhou Academy of Agricultural Sciences were used as materials. The genotyping-by-sequencing(GBS) was used to detect the single nucleotide polymorphism(SNP) sites in their genomes, and the genetic characteristics of the 59 Sect. Thea(L.) Dyer resources were analyzed based on the obtained high-quality genomic SNP sites. 【Result】A total of 45.84 Gb clean reads were obtained from the 59 Sect. Thea (L.) Dyer resources, with an average of 795.6 Mb per mate-rial, accounting for 26.5% of the improved version of tea reference genome (2.93 Gb), and the average alignment rate was 72.62%. A total of 248772 high-quality genomic SNPs were obtained, of which 83.98% and 16.02% were distributed in intergenic region and genic region, respectively. There were 22614 SNP sites distributed in introns, 15038 SNP sites distributed in exon regions, and 2203 SNP sites distributed in untranslated regions(UTR). The results showed that the observed heterozygosity(Ho) of them ranged from 0.016 to 0.081, the expected heterozygosity(He) was 0.006 to 0.064, and F value was -0.331 to 0.737. The 59 samples were divided into three groups based on principal component analysis (PCA),phylogenetic tree construction and genetic structure analysis. Among them,all of the Camellia sinensis were in one group,C. remoterrata and C. tachangensis were in one group,and the nine C. costata were in one group. However,C. remoterrata and C. tachangensis and the C. tachangensis from two different regions could be further divided into different independent subgroups,and the C. sinensis var. assamica(CSA) and C. sinensis var. sinensis(CSS) were also in the diffe-rent branches of evolutionary tree. Additionally,based on the phylogenetic tree,the genetic relationships between C. costata and C. remoterrata or C. tachangensis were more closer to that between C. costata and C. sinensis. The gene structure analysis showed that there was a fusion genetic background among the four classifications of Sect. Thea(L.) Dyer resources(C. sinensis, C. remoterrata, C. tachangensis and C. costata) used in this study. According to geographical origin and germplasm type, the 59 Sect. Thea (L.) Dyer resources were divided into 11 groups, and the gene flow analysis showed that there was a low level of gene flow among different groups and a high level of gene flow within the group. 【Conclusion】There are obvious differences among different Sect. Thea (L.) Dyer groups at the genomic level. Based on the gene-tic structure,C. tachangensis and C. remoterrata are more appropriate to be divided into two varieties, although they are combined in the classical morphological classification. The intraspecific genetic relationship of different Sect. Thea(L.) Dyer is directly related to their geographical origin. Therefore, the same geographical source material hybrid application should be avoided in breeding practice.
Key words:Sect. Thea(L.) Dyer; Camellia sinensis; SNP; genome; phylogenetic evolution; genetic structure; gene flow
Foundation item:Science and Technology Support Plan Project of Guizhou Province(QKHZC〔2019〕2254);Guizhou Academy of Agricultural Sciences High Value Patent Cultivation Project(QNKYZLPX〔2018〕02)
0 引言
【研究意義】贵州地处我国西南,独特的地理位置和得天独厚的气候因素孕育了丰富的茶组[Sect. Thea(L.) Dyer]植物资源(陈正武等,2009;鄢东海,2009)。近年来,随着贵州省茶产业的发展,茶树资源乃至茶组植物资源的保护、开发和利用引起了人们的高度重视,其主要原因是这些丰富的茶组植物资源不仅是物种多样性的象征,同时为研究茶组植物资源的分类、进化、驯化及育种利用提供了宝贵的遗传材料(马建强等,2015)。除了作为茶饮原料主要来源而为人熟知的茶种[Camellia sinensis(L.) O. Kuntze]外,茶组内部还存在大量的不同种和变种类型(汪云刚等,2010)。由于茶组植物内部种和变种的多样性及茶组植物本身的异花传粉特性,致使其成为植物分类学上最复杂的物种之一(张宏达,1984;陈亮等,2000;闵天禄,2000),给茶组植物的高效保护和利用带来巨大障碍。了解和掌握茶组植物间的遗传关系是对其进行保护和利用的前提,但目前针对茶组植物分类研究主要依托于我国西南地区茶组植物资源的调查和考察(陈亮等,2000;汪云刚等,2010)。因此,开展贵州地方茶组植物资源的遗传关系探究不仅有助于建立高效的分类保护和利用机制,还能为茶组植物的分类研究提供参考。【前人研究进展】长期以来针对茶组植物亲缘关系及其遗传多样性的分析主要基于表型性状和生化组分,但受茶组植物本身的自交不亲和、生育期长、遗传背景复杂及分布环境恶劣等因素的影响,仅依靠传统的经典形态学已难以达到研究目的。随着分子生物学技术的发展,基于DNA序列多态性的RFLP、RAPD、AFLP、ISSR、SSR和SNP等类型的分子标记因其稳定性、准确性和高效性,已发展成为茶组植物资源鉴定最广泛的技术手段(Fang et al.,2014;Tan et al.,2015;Liu et al.,2017;Meegahakumbura et al.,2017)。Yao等(2011)基于EST-SSR分子标记对来自我国14个省(区)的450份茶树种质进行遗传多样性和群体结构分析,结果表明云南、贵州和广西的茶树资源具有较高的遗传多样性。牛素珍等(2012)利用EST-SSR分子标记对贵州代表性的25个地方茶树群体种和3个育成品种进行遗传多样性和亲缘关系分析,结果发现贵州地方茶树品种间遗传差异明显,黔南地区的茶树资源遗传多样性程度明显高于黔北地区,且两地区间的基因交流频率较低。Tan等(2015)利用SSR分子标记对来自我国13个省(区)的128个优良无性系茶树品种进行基因分型分析,结果发现有29个品种的母本或父本为福鼎大白茶。上述研究虽然从根本上加深了人们对茶树资源遗传关系的认识,但由于分子标记数量的限制及参考基因组的缺乏,现有的分子标记很难覆盖整个基因组。茶树基因组测序的完成(Xia et al.,2017,2020;Wei et al.,2018;Zhang et al.,2020a)为从全基因组水平上解析茶树乃至茶组植物的起源进化、遗传结构和亲缘关系提供了极大便利。基于茶树参考基因组的简化基因组测序和基因组重测序等技术已成功应用于茶树不同变种间及不同种质类型间的遗传关系解析(Yang et al.,2016;郭燕等,2019;乔大河等,2019;Niu et al.,2019;An et al.,2020)。【本研究切入点】目前基于全基因组SNP的茶组植物遗传关系分析主要集中于茶种及变种间,针对茶种与茶组其他种间及茶组其他种内遗传关系的研究鲜见报道。【拟解决的关键问题】以从贵州不同区域收集的茶组植物资源及贵州省茶叶研究所茶树种质资源圃保存的不同省(市)育成茶树品种(系)为材料,利用GBS(Genoty-ping by sequencing)简化基因组测序技术进行基因组SNP变异检测,并分析不同茶组植物的亲缘关系,为初步解晰基于形态分类学的经典分类法的不同茶组植物类别在基因组水平上的遗传关系,为贵州地方茶组植物的保护、利用打下基础,同时为茶组植物的分类和起源进化研究提供参考。
1 材料与方法
1. 1 研究材料
供试茶组植物材料共59份,其中,18份来自贵州省茶叶研究所茶树种质资源圃,包括贵州省内育成品种材料10份(黔茶1号、黔茶7号、黔茶8号、黔湄601、黔湄701、黔辐4号、黔湄419、鸟王种、贵茶育8号和单株1号)、省外育成品种8份(福鼎大白茶、金观音、台茶12、紫娟、龙井43、槠叶齐、保靖黄金茶和名山131),其他41份茶组植物材料采自贵州省内不同县区,包括取自花溪区(7份)、沿河县(5份)和都匀市(5份)的茶种(C. sinensis)材料17份,取自务川县的疏齿茶(C. remotiserrata)材料5份,取自普安县和兴义市的大厂茶(C. tachangensis)材料各5份及取自三都县的突肋茶(C. costata)材料9份。对上述材料的新生枝条取样插入花泥,带回实验室后将幼嫩叶片用液氮速冻后放入-80 ℃冰箱保存备用。根据茶组类别、种质类型和地理来源,可将这些材料分为9组(A~I),详见表1。主要仪器设备:伯乐核酸电泳仪(Bio-Rad,美国)、伯乐凝胶成像仪(Bio-Rad,美国)和NanoDrop ND-2000分光光度计(Thermo,美国)。
1. 2 试验方法
1. 2. 1 基因组DNA提取及GBS测序文库构建 基因组DNA提取采用改良的CTAB法,经1.5%琼脂糖凝胶电泳检测后送至武汉贝纳科技服务有限公司进行GBS测序及文库构建:质检合格的DNA首先用限制性内切酶Sac I和Mse I进行双酶切,然后将酶切片段连接上设计好的测序接头,对连接上接头的片段进行回收纯化后,选择插入片段长度在370~420 bp的DNA片段进行富集扩增,利用Illumina HiSeq测序平台基于双末端150 bp进行上机测序。
1. 2. 2 基因组SNP鉴定 测序获得的原始图像数据首先经碱基识别(Base calling)分析转化为FASTQ文件格式的原始数据(Raw reads),然后利用Trimmomatic对原始数据进行质控过滤(Bolger et al.,2014),去除含接头序列和低质量序列,包括序列两端碱基质量低于10的碱基及以4个碱基为窗口进行滑动、窗口内碱基平均质量小于15的序列,同时过滤掉序列长度小于50的序列,最终获得高质量序列(Clean reads)。以茶树品种舒茶早改良版基因组(Xia et al.,2019)为参考,将获得的高质量序列分别比对茶树参考基因组获得bam格式文件,然后利用GATK以HaplotypeCaller为参数进行变异检测(McKenna et al.,2010),同时对变异检测结果进行过滤,包括过滤掉质量值小于50的变异位点、QD<2.0||ReadPosRankSum<-8.0||FS>60.0||MLEAF>0.05的位点及Miss>0.5的位点,最终获得高质量的SNP位点,并利用SnpEff对SNP位点类型进行注释分析(Cingolani et al.,2012)。
1. 2. 3 群體进化树构建、遗传结构及主成分分析
基于获得的高质量SNP位点对59份茶组植物材料进行遗传关系分析:利用Plink进行茶组植物样品的杂合度分析(Purcell et al.,2007);利用PHYLIP(https://evolution.genetics.washington.edu/phylip.html)基于默认参数以邻接法(Neighbor-joining,NJ)构建系统发育进化树,并利用iTOL在线网站(https://itol.embl.de/)进行可视化;利用ADMIXTURE按照默认参数进行种群结构分析(Alexander et al.,2009),K取值1~7,基于最小交叉熵确定最佳分群数;利用EIGENSOFT基于默认参数进行主成分分析(Patterson et al.,2006),并利用R语言绘制主成分分布图;利用BayesAss(http://www.rannala.org/?page_id=245)基于SNP信息估算不同茶组群体间基因流(Mussmann et al.,2019)。
2 结果与分析
2. 1 茶组植物的测序质量及SNP位点鉴定结果
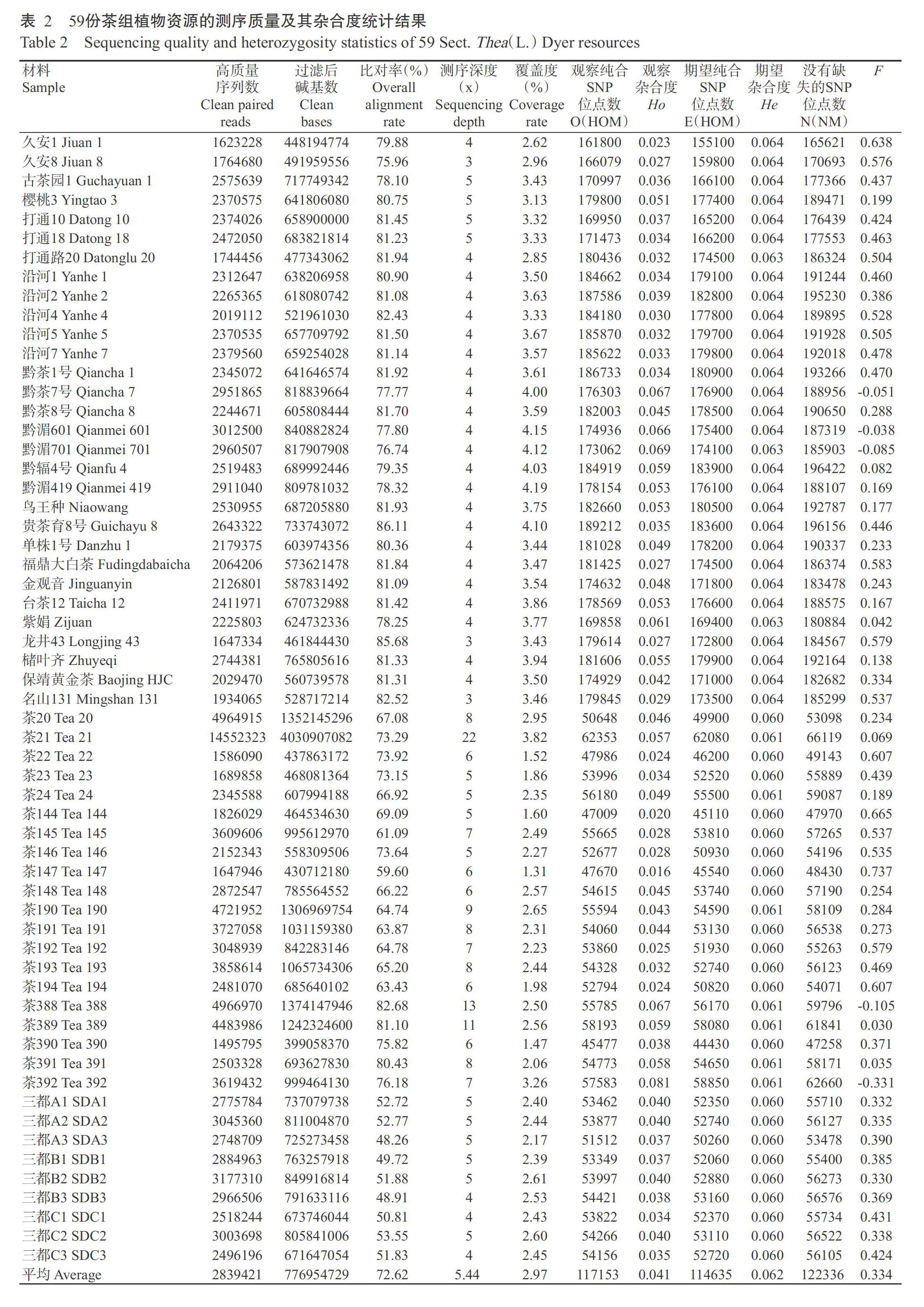
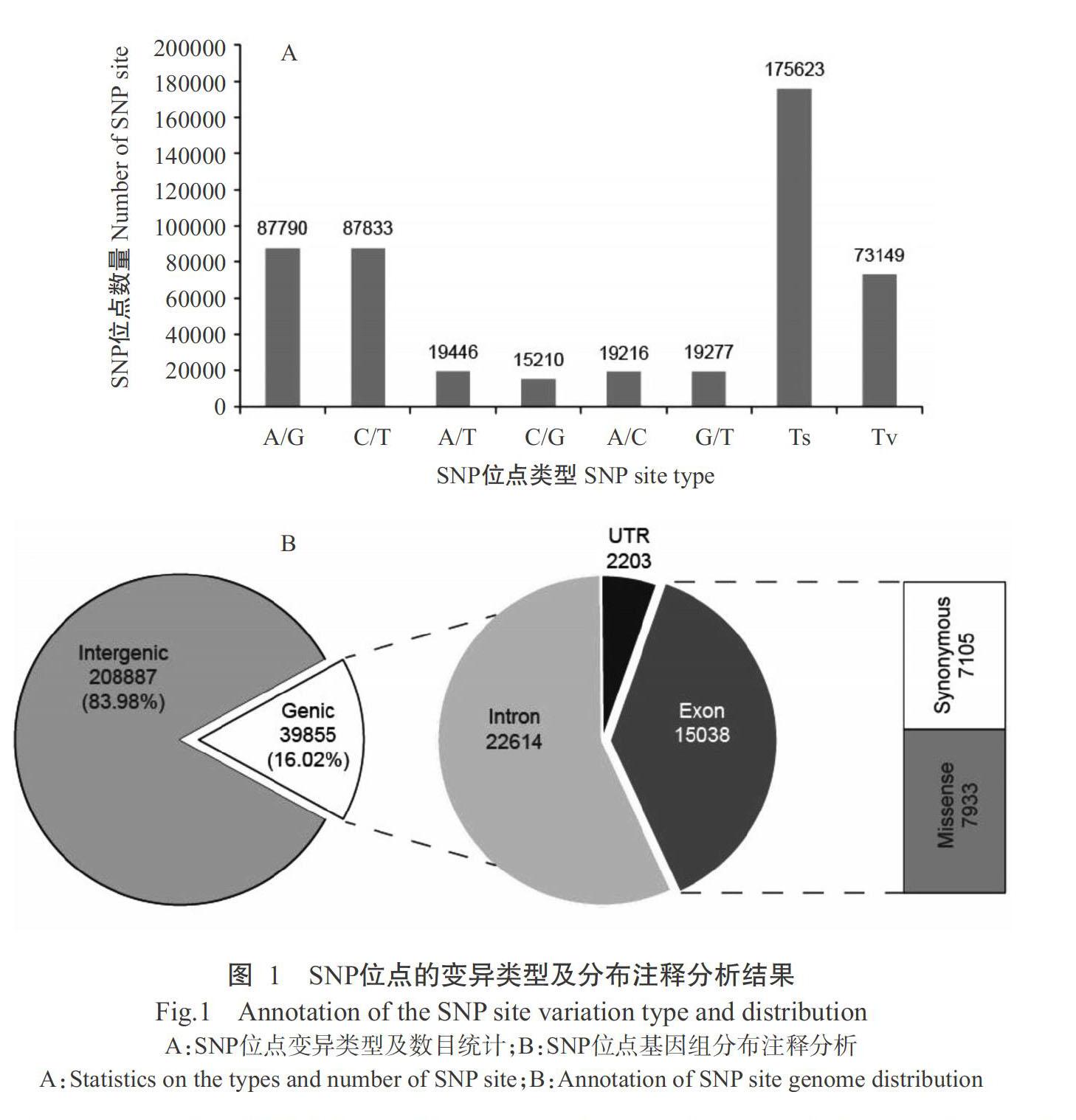
59份茶组植物材料共获得50.34 Gb测序数据,过滤掉低质量数据后最终获得45.84 Gb高质量序列数据,平均每个材料获得2839421条成对的高质量序列,平均每个材料的高质量序列数据为795.6 Mb,约占改良版茶树基因组大小(2.93 Gb)的26.5%。将过滤后的序列比对改良版茶树基因组,结果发现59份茶组植物材料的平均比对率为72.62%,且茶种(C. sinensis)材料的平均比对率(80.50%)远高于其他茶组类别材料的平均比对率(61.10%),说明不同茶组植物基因组水平上存在差异(表2)。利用GATK对每个材料比对到参考基因组的序列进行变异检测,经过滤后得到256226个高质量SNP位点和InDel位点,其中SNP位点248772个,InDel位点7454个。鉴于2种变异位点数量的差异,利用SNP位点进行后续分析。对248772个高质量SNP位点的变异类型进行分析,结果显示转换类型(Ts,A/G或C/T)的SNP位点最多,约占70.6%,颠换类型(Tv)的SNP位点约占29.1%,Ts和Tv的比值为2.4(图1-A)。基因结构分布注释结果显示,83.98%的高质量SNP位点分布在基因间区(Intergenic),16.02%分布在基因区(Genic);对基因区的SNP位点进一步分析,结果显示有22614个SNP位点分布在内含子(Intron)、15038个SNP位点分布在外显子区(Exon)、2203个SNP位点分布在非翻译区(UTR);分布于外显子区的SNP位点中有7105个导致同义突变(Synonymous),另外7933个SNP位点导致非同义变异(Missense)(图1-B)。
由表2还可知,59份供试材料的观察杂合度(Ho)为0.016~0.081,期望杂合度(He)为0.060~0.064。基于Plink计算得出F,用于衡量杂合度,F越大,杂合度越小,反之亦然。F为-0.331~0.737,说明不同茶组植物材料的杂合度存在明显差异,除了大多数育成茶树品种具有较高的杂合度外,部分古茶树资源(如茶388~茶392)同样表现出较高的杂合度,甚至高于育成品种;大多数野生茶树的杂合度低于育成茶树品种的杂合度。18份育成茶树品种中,黔湄701、黔茶7号和黔湄601的杂合度较高,龙井43、福鼎大白茶和名山131的杂合度较低(表2),可能与前三者是从杂交后代选育,而后三者是从地方群体种中选育有关。
2. 2 茶组植物的主成分分析结果
利用获得的高质量SNP位点对59份茶组植物材料进行基于主成分分析的亲缘关系鉴定,结果如图2所示。根据第一主成分(PC1)和第二主成分(PC2)可将59份茶组植物材料划分为3个类群,其中全部茶种(A、B、C、D和H分组)材料归在一个类群,大部分疏齿茶(E分组)和大厂茶(F分组)材料归在一个类群,9份突肋茶(即I分组)材料单独在一个类群,但少部分大厂茶、疏齿茶材料和茶种材料出现混群。
2. 3 茶组植物的遗传结构分析结果
由图3-A可知,茶种、疏齿茶、大厂茶和突肋茶4类茶组植物分别处于4个独立分支上,同时由于大厂茶与突肋茶的亲缘关系较近,可划分为一个类群,与主成分分析结果高度相似。在部分已知亲缘关系的茶种材料中,黔辐4号是黔湄419的辐照种子后代,二者在紧密相连的同一个分支上;黔茶7号、黔辐4号、黔湄419、紫娟、黔湄601和黔湄701均属于茶种中的阿萨姆变种(C. sinensis var. assamica,CSA)也聚在一起,而与中国变种(C. sinensis var. sinensis,CSS)(福鼎大白茶、黔茶1号、黔茶8号、金观音、鸟王种、名山131、槠叶齐、贵茶育8号、单株1号、龙井43等)处在不同的亚支上。上述结果一方面证实系统发育进化树的可靠性,另一方面表明茶种的变种间及不同茶组植物间在基因组水平上均存在明显的遗传差异。来自兴义和普安的大厂茶材料(茶144~茶148和茶190~茶194)虽然聚在同一个分支上,但这10份材料可依据地理来源进一步分为2个分支,表明地理阻隔导致大厂茶种群间遗传差异;但对于茶种材料来说,其亲缘关系的远近与材料地理来源并未显示出一致性,如来自湖南的保靖黄金茶与来自贵州久安的古茶园1及来自贵州沿河的沿河5具有相同的进化分支,来自我国台湾的台茶12与来自浙江的龙井43及来自贵州的贵茶育8号聚在一起,可能与茶种经受更多的人工驯化有关。
为进一步明确不同茶组植物材料间的遗传背景关系,利用ADMIXTURE对其遗传结构进行分析,结果显示,在K=3时,交叉熵值最小,表明根据59份茶组植物材料的遗传结构,其最佳分群数为3个,与主成分分析结果和系统发育进化树构建情况一致。如图3-B所示,在K=3时,3个类群呈现出独特的遗传背景,且表明疏齿茶和大厂茶亲缘关系很近,值得注意的是疏齿茶、大厂茶和茶种三者的遗传结构中均呈现出突肋茶的遗传背景;在K=4时,疏齿茶和大厂茶呈现出不同的遗传结构,但茶种和大厂茶种质材料中仍存在突肋茶的遗传背景。
2. 4 茶组植物的基因流分析结果
由表2可知,根据茶组植物材料的地理来源和种质类型将59份资源分为9组(A~I),其中I组的突肋茶根据地理来源又可分为3个亚组(I-A、I-B和I-C),共计11个种群。基于SNP位点利用BayesAss分析11个种群内及种群间的基因流,结果如表3所示。不同种群间存在较低水平的基因流(0.002~0.056),除种群E(疏齿茶)向种群A(花溪古茶树)和种群H(都匀古茶树)的基因流大于0.050之外,其他种群间的基因流均小于0.050。种群内基因流均在0.700以上,表明种群内部具有较高的基因流。
3 讨论
SNP分子标记作为物种基因组中最丰富的变异类型,其全基因组的覆盖广是其他分子标记无可比拟的优势(黎裕等,2015)。茶组植物作为变异类型最丰富、分类最复杂的物种之一,其不同种及变种间的遗传关系尚不清楚。基于全基因组SNP位点对茶组植物中茶种的遗传关系进行鉴定,但对于除茶种之外其他茶组植物种类的遗传关系鲜见报道。本研究通过简化基因组测序及比对茶树基因组发现,无论是野生茶树资源还是栽培型古茶树资源均具有较高的比对率,而突肋茶、大厂茶和疏齿茶的比对率远低于茶种,尤其是9份突肋茶的比对率均在55.00%以下,表明这4类茶组植物类别在基因组水平上存在差异。而基于基因组SNP位点的主成分分析结果显示,59份茶组植物材料划分为3个类群,不同类群间存在明显差异,虽然大厂茶和疏齿茶处在同一个类群,但二者还可进一步分为不同的亚群。这些结果在系统发育进化树分析和遗传结构分析结果中均得到证实,与Yang等(2016)对于大理茶(C. taliensis)、厚轴茶(C. crassicolumna)和茶三者的遗传关系分析结果基本一致,表明不同茶组植物种类间除了表型的差异外,其基因型也存在明显差异。从分类角度上来看,大厂茶最明显的特征是子房5室、花柱5裂、子房、顶芽、幼枝、叶柄均无毛;疏齿茶最明显特征为子房存在5-4-3室变异但以4-5室为主、子房无毛但顶芽有毛;突肋茶则为子房3室、子房无毛、顶芽无毛,而茶种为子房3室、子房有毛、幼枝有毛(闵天禄,2000)。根据表型特征,闵天禄(2000)将大厂茶和疏齿茶归为大厂茶的2个变种,而陈亮等(2000)在后续分类中直接将疏齿茶归为大厂茶。但本研究认为,将大厂茶和疏齿茶分为2个变种更合适。此外,一般认为茶组植物的演化是子房室数逐渐减少,子房无毛到有毛(陈亮等,2000)。本研究遗传结构分析结果显示,突肋茶、大厂茶、疏齿茶和茶种的遗传背景中均存在彼此融合现象,表明其在进化上具有共同的起源。而基于上述演化路线,作为5室茶系的大厂茶和疏齿茶在进化上要早于3室茶系的突肋茶和茶种,但系统发育进化树显示突肋茶与大厂茶和疏齿茶的亲缘关系较其与茶种近,可能是突肋茶较茶种保留了更多的祖先茶组植物性状。另外,从表型性状来看,突肋茶与茶种的最大分类区别在于子房有无茸毛,暗示茶组植物子房茸毛性状的差异在分类依据上要优先于子房室数的差异,后续研究应扩大样品量进行深入探究。
茶种中的CSA和CSS是茶叶生产上的主要栽培类型,二者除了叶片大小和树型的明显差异外,其基因组水平差异也得到广泛报道(Meegahakumbura et al.,2017;An et al.,2020;Wang et al.,2020;Xia et al.,2020)。Yu等(2020)基于转录组SNP位点对分布于我国几乎所有产茶省(区)的136份茶树资源(其中有128份为育成品种)进行群体划分,结果显示CSA和CSS具有明显的种群差异,但其材料的地理差异并未导致种群差异。本研究同樣发现已知的CSA(黔茶7号、黔辐4号、黔湄419、黔湄601、紫娟、黔湄701)和CSS(福鼎大白茶、黔茶1号、黔茶8号、金观音、鸟王种、名山131、槠叶齐、贵茶育8号、单株1号、龙井43等)呈现不同的进化分支,而来自不同省市的育成品种甚至是省外的育成品种与贵州本地的古茶树资源处在相同的进化分支上,表明茶种间的差异主要是由于茶种类型不同,且茶种内的差异远小于茶种间的差异。虽然来自不同地区的茶树资源聚在相同分支上,而在分支内部来自相同地区的材料间亲缘关系更近,与基因流分析结果相一致。这种现象在姚明哲等(2012)对川、渝地方茶树品种群体结构分析、郭燕等(2019)对贵州久安古茶树遗传关系分析中均被发现,且在本研究中来自贵州兴义和普安的大厂茶材料间也表现出同样的现象,说明地理来源虽然不是导致茶组植物遗传分化的主要因素,但与种内亲缘关系直接相关。因此在育种实践中尽量避免相同地理来源材料间的杂交应用。
此外,本研究发现除了大多数育成茶树品种具有较高的杂合度外,部分古茶树资源同样表现出较高的杂合度,甚至高于育成品种,而野生茶树的杂合度整体上低于育成茶树品种的杂合度,与Yang等(2016)对18份茶组植物材料杂合性分析结果一致。一般而言,相比于野生资源,栽培种经历了人工选择进而导致其遗传多样性降低(Varshney et al.,2019)。Niu等(2019)通过构建415份茶组植物材料(包含栽培茶树品种、古茶树和野生茶树)的系统发育进化树,结果发现古茶树的遗传多样性要高于野生茶树;Zhang等(2020b)认为这可能是野生茶树取样不全面或混有栽培种的原因。本研究对大厂茶和疏齿茶遗传关系的探讨同样存在样本量少的局限性。从遗传进化角度来看,植物的交配系统、选择、遗传漂移及突变和迁移均是影响植物群体遗传结构的进化因子,而遗传漂移又是生物小群体固有的属性(徐刚标,2009)。本研究中,虽然大厂茶和疏齿茶均具有独立的进化分支,但其分别来自不同区域,难以排除地域差异导致的遗传差异。因此,在后续茶组植物种质评价过程中应适度增加野生茶树(或茶组植物)资源的数量占比及扩大地理区域,以准确评价其遗传特性。
4 结论
不同茶组植物在基因组水平上存在明显差异,经典形态学分类上合二为一的大厂茶和疏齿茶在遗传结构上存在差异,即分为2个变种更合适;茶组植物种内亲缘关系与其地理来源直接关系,故在育种实践中应尽量避免相同地理来源材料间的杂交应用。
参考文献:
陈亮,虞富莲,童启庆. 2000. 关于茶组植物分类与演化的讨论[J]. 茶叶科学,20(2):89-94. doi:10.13305/j.cnki.jts. 2000.02.003. [Chen L,Yu F L,Tong Q Q. 2000. Discussions on phylogenetic classification and evolution of Sect. Thea[J]. Journal of Tea Science,20(2):89-94.]
陈正武,刘红梅,曹雨. 2009. 贵州野生茶树资源及地方品种变异类型的保护与利用[J]. 贵州农业科学,37(7):188-190. doi:10.3969/j.issn.1001-3601.2009.07.062. [Chen Z W,Liu H M,Cao Y. 2009. Protection and utilization of variation type of wild tea resources and local tea varieties in Guizhou[J]. Guizhou Agricultural Sciences,37(7):188-190.]
郭燕,喬大河,杨春,李燕,陈正武,陈娟. 2019. 基于全基因组SNP的贵州久安古茶树遗传关系分析[J]. 植物遗传资源学报,20(1):26-36. doi:10.13430/j.cnki.jpgr.20180529001. [Guo Y,Qiao D H,Yang C,Li Y,Chen Z W,Chen J. 2019. Genetic diversity of old tea plant resources in Jiuan City of Guizhou Province,using genome-wide SNP[J]. Journal of Plant Genetic Resources,20(1):26-36.]
黎裕,李英慧,杨庆文,张锦鹏,张金梅,邱丽娟,王天宇. 2015. 基于基因组学的作物种质资源研究:现状与展望[J]. 中国农业科学,48(17):3333-3353. doi:10.3864/j.issn.0578-1752.2015.17.003. [Li Y,Li Y H,Yang Q W,Zhang J P,Zhang J M,Qiu L J,Wang T Y. 2015. Geno-mics-based crop germplasm research:Advances and perspectives[J]. Scientia Agricultura Sinica,48(17):3333-3353.]
马建强,姚明哲,陈亮. 2015. 茶树种质资源研究进展[J]. 茶叶科学,35(1):11-16. doi:10.13305/j.cnki.jts.2015.01.003. [Ma J Q,Yao M Z,Chen L. 2015. Research progress on germplasms of tea plant(Camellia sinensis)[J]. Journal of Tea Science,35(1):11-16.]
闵天禄. 2000. 世界山茶属的研究[M]. 昆明:云南科技出版社. [Ming T L. 2000. Studies on Camellia in the world[M]. Kunming:Yunnan Science and Technology Press.]
牛素贞,刘玉倩,杨锦标,樊卫国. 2012. 贵州茶树地方品种的EST-SSR遗传多样性分析[J]. 浙江农业学报,24(5):836-841. doi:10.3969/j.issn.1004-1524.2012.05.016. [Niu S Z,Liu Y Q,Yang J B,Fan W G. 2012. Analysis of genetic diversity of tea landrace resources in Guizhou by EST-SSR markers[J]. Acta Agriculturae Zhejiangensis,24(5):836-841.]
乔大河,郭燕,杨春,李燕,陈娟,陈正武. 2019. 贵州省主要栽培茶树品种指纹图谱构建与遗传结构分析[J]. 植物遗传资源学报,20(2):412-425. doi:10.13430/j.cnki.jpgr. 20180801002. [Qiao D H,Guo Y,Yang C,Li Y,Chen J,Chen Z W. 2019. Fingerprinting construction and genetic structure analysis of the main cultivated tea varieties in Guizhou Province[J]. Journal of Plant Genetic Resources,20(2):412-425.]
汪云刚,刘本英,宋维希,马玲,蒋会兵,矣兵,唐一春,李友勇,孙雪梅,王平盛. 2010. 云南茶组植物的分布[J]. 西南农业学报,23(5):1750-1753. doi:10.16213/j.cnki.scjas.2010.05.043. [Wang Y G,Liu B Y,Song W X,Ma L,Jiang H B,Yi B,Tang Y C,Li Y Y,Sun X M,Wang P S. 2010. Distribution of Sect. Thea(L.) Dyer in Yunnan Pro-vince[J]. Southwest China Journal of Agricultural Scien-ces,23(5):1750-1753.]
徐剛标. 2009. 植物群体遗传学[M]. 北京:科学出版社. [Xu G B. 2009. Plant population genetics[M]. Beijing:Science Press.]
鄢东海. 2009. 贵州茶树种质资源研究进展及野生茶树资源调查[J]. 贵州农业科学,37(7):184-187. doi:10.3969/j.issn. 1001-3601.2009.07.061. [Yan D H. 2009. Research progress on tea germplasm resources and investigation of wild tea resource in Guizhou[J]. Guizhou Agricultural Scien-ces,37(7):184-187.]
姚明哲,马春雷,金基强,马建强,陈亮. 2012. 川、渝地方茶树品种的遗传多样性和群体结构[J]. 茶叶科学,32(5):419-425. doi:10.13305/j.cnki.jts.2012.05.011. [Yao M Z,Ma C L,Jin J Q,Ma J Q,Chen L. 2012. Genetic diversity and population structure of local tea landraces in Sichuan and Chongqing revealed by EST-SSR markers[J]. Journal of Tea Science,32(5):419-425.]
张宏达. 1984. 茶叶植物资源的订正[J]. 中山大学学报(自然科学版),(1):1-12. [Chang H T. 1984. A revision on the tea resource plants[J]. Acta Scientiarum Naturalium Universitatis Sunyatsen,(1):1-12.]
Alexander D H,Novembre J,Lange K. 2009. Fast model-based estimation of ancestry in unrelated individuals[J]. Genome Research,19(9):1655-1664. doi:10.1161/01.ATV. 0000137190.63214.c5.
An Y L,Mi X Z,Zhao S Q,Guo R,Xia X B,Liu S R,Wei C L. 2020. Revealing distinctions in genetic diversity and adaptive evolution between two varieties of Camellia sinensis by whole-genome resequencing[J]. Frontier in Plant Science,11:603819. doi:10.3389/fpls.2020.603819.
Bolger A M,Lohse M,Usadel B. 2014. Trimmomatic:A flexible trimmer for Illumina sequence data[J]. Bioinforma-tics,30(15):2114-2120. doi:10.1093/bioinformatics/btu 170.
Cingolani P,Platts A,Wang L L,Coon M,Nguyen T,Wang L,Land S J,Lu X Y,Ruden D M. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms,SnpEff:SNPs in the genome of Droso-phila melanogaster strain w1118;iso-2;iso-3[J]. Fly(Austin),6(2):80-92. doi:10.4161/fly.19695.
Fang W P,Meinhardt L W,Tan H W,Zhou L,Mischke S,Zhang D. 2014. Varietal identification of tea(Camellia sinensis) using nanofluidic array of single nucleotide polymorphism(SNP) markers[J]. Horticulture Research,1:14035. doi:10.1038/hortres.2014.35.
Liu S R,Liu H W,Wu A L,Hou Y,An Y L,Wei C L. 2017. Construction of fingerprinting for tea plant(Camellia sinensis) accessions using new genomic SSR markers[J]. Molecular Breeding,37(8):93. doi:10.1007/s11032-017-0692-y.
McKenna A,Hanna M,Banks E,Sivachenko A,Cibulskis K,Kernytsky A,Garimella K,Altshuler D,Gabriel S,Daly M,DePristo M A. 2010. The genome analysis toolkit:A MapReduce framework for analyzing next-generation DNA sequencing data[J]. Genome Research,20(9):1297-1303. doi:10.1101/gr.107524.110.
Meegahakumbura M K,Wambulwa M C,Li M M,Thapa K K,Sun Y S,Moller M,Xu J C,Yang J B,Liu J,Liu B Y,Li D Z,Gao L M. 2017. Domestication origin and bree-ding history of the tea plant(Camellia sinensis) in China and India based on nuclear microsatellites and cpDNA sequence data[J]. Frontiers in Plant Science,8:2270. doi:10.3389/fpls.2017.02270.
Mussmann S M,Douglas M R,Chafin T K,Douglas M E. 2019. BA3-SNPs:Contemporary migration reconfigured in BayesAss for next generation sequence data[J]. Me-thods in Ecology and Evolution,10:1808-1813. doi:10.1111/ 2041-210X.13252.
Niu S Z,Song Q F,Koiwa H,Qiao D H,Zhao D G,Chen Z W,Liu X,Wen X P. 2019. Genetic diversity,linkage disequilibrium,and population structure analysis of the tea plant(Camellia sinensis) from an origin center,Guizhou plateau,using genome-wide SNPs developed by genoty-ping-by-sequencing[J]. BMC Plant Biology,19(1):328. doi:10.1186/s12870-019-1917-5.
Patterson N,Price A L,Reich D. 2006. Population structure and eigenanalysis[J]. PLoS Genetics,2(12):e190. doi:10.1371/journal.pgen.0020190.
Purcell S,Neale B,Todd-Brown K,Thomas L,Ferreira M A,Bender D,Maller J,Sklar P,de Bakker P I,Daly M J,Sham P C. 2007. PLINK:A tool set for whole-genome association and population-based linkage analyses[J]. Ame-rican Journal of Human Genetics,81(3):559-575. doi:10.1086/519795.
Tan L Q,Peng M,Xu L Y,Wang L Y,Chen S X,Zou Y,Qi G N,Cheng H. 2015. Fingerprinting 128 Chinese clonal tea cultivars using SSR markers provides new insights into their pedigree relationships[J]. Tree Genetics & Genomes,11(5):90. doi:10.1007/s11295-015-0914-6.
Varshney R K,Thudi M,Roorkiwal M,He W,Upadhyaya H D,Yang W,Bajaj P,Cubry P,Rathore A,Jian J,Doddamani D,Khan A W,Garg V,Chitikineni A,Xu D,Gaur P M,Singh N P,Chaturvedi S K,Nadigatla G V P R,Krishnamurthy L,Dixit G P,Fikre A,Kimurto P K,Sreeman S M,Bharadwaj C,Tripathi S,Wang J,Lee S H,Edwards D,Polavarapu K K B,Penmetsa RV,Crossa J,Nguyen H T,Siddique K H M,Colmer T D,Sutton T,von Wettberg E,Vigouroux Y,Xu X,Liu X. 2019. Resequencing of 429 chickpea accessions from 45 countries provides insights into genome diversity,domestication and agronomic traits[J]. Nature Genetics,51(5):857-864. doi:10.1038/s41588-019-0401-3.
Wang X C,Feng H,Chang Y X,Ma C L,Wang L Y,Hao X Y,Li A L,Cheng H,Wang L,Cui P,Jin J Q,Wang X B,Wei K,Ai C,Zhao S,Wu Z C,Li Y Y,Liu B Y,Wang G D,Chen L,Ruan J,Yang Y. 2020. Population sequencing enhances understanding of tea plant evolution[J]. Nature Communications,11(1):4447. doi:10.1101/2020.03.19. 998393.
Wei C L,Yang H,Wang S B,Zhao J,Liu C,Gao L P,Xia E H,Lu Y,Tai Y L,She G B,Sun J,Cao H S,Tong W,Gao Q,Li Y Y,Deng W W,Jiang X L,Wang W Z,Chen Q,Zhang S H,Li H J,Wu J L,Wang P,Li P H,Shi C Y,Zheng F Y,Jian J B,Huang B,Shan D,Shi M M,Fang C B,Yue Y,Li F D,Li D X,Wei S,Han B,Jiang C J,Yin Y,Xia T,Zhang Z Z,Bennetzen J L,Zhao S C,Wan X C. 2018. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality[J]. Proceedings of the National Academy of Sciences of the United States of America,115(18):E4151-E4158. doi:10.1073/pnas.1719 622115.
Xia E H,Zhang H B,Sheng J,Li K,Zhang Q J,Kim C,Zhang Y,Liu Y,Zhu T,Li W,Huang H,Tong Y,Nan H,Shi C,Shi C,Jiang J J,Mao S Y,Jiao J Y,Zhang D,Zhao Y,Zhao Y J,Zhang L P,Liu Y L,Liu B Y,Yu Y,Shao S F,Ni D J,Eichler E E,Gao L Z. 2017. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis[J]. Molecular Plant,10(6):866-877. doi:10.1016/j.molp.2017.04.002.
Xia E H,Li F D,Tong W,Yang H,Wang S B,Zhao J,Liu C,Gao L P,Tai Y L,She G B,Sun J,Cao H S,Gao Q,Li Y Y,Deng W W,Jiang X L,Wang W Z,Chen Q,Zhang S H,Li H J,Wu J L,Wang P,Li P H,Shi C Y,Zheng F Y,Jian J B,Huang B,Shan D,Shi M M,Fang C B,Yue Y,Wu Q,Ge R H,Zhao H J,Li D X,Wei S,Han B,Jiang C J,Yin Y,Xia T,Zhang Z Z,Zhao S C,Bennetzen J L,Wei C L,Wan X C. 2019. The tea plant reference genome and improved gene annotation using long-read and paired-end sequencing data[J]. Scientific Data,6(1):122. doi:10.1038/s41597-019-0127-1.
Xia E H,Tong W,Hou Y,An Y L,Chen L B,Wu Q,Liu Y L,Yu J,Li F D,Li R P,Li P H,Zhao H J,Ge R H,Huang J,Mallano A I,Zhang Y R,Liu S R,Deng W W,Song C K,Zhang Z L,Zhao J,Wei S,Zhang Z Z,Xia T,Wei C L,Wan X C. 2020. The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into genome evolution and adaptation[J]. Mole-cular Plant,13(7):1013-1026. doi:10.1016/j.molp.2020. 04.010.
Yang H,Wei C L,Liu H W,Wu J L,Li Z G,Zhang L,Jian J B,Li Y Y,Tai Y L,Zhang J,Zhang Z Z,Jiang C J,Xia T,Wan X C. 2016. Genetic divergence between Camellia sinensis and its wild relatives revealed via genome-wide SNPs from RAD sequencing[J]. PLoS One,11(3):e015 1424. doi:10.1371/journal.pone.0151424.
Yao M Z,Ma C L,Qiao T T,Jin J Q,Chen L. 2011. Diversity distribution and population structure of tea germplasms in China revealed by EST-SSR markers[J]. Tree Genetics & Genomes,8(1):205-220. doi:10.1007/s11295- 011-0433-z.
Yu X M,Xiao J J,Chen S,Yu Y,Ma J Q,Lin Y Z,Li R Z,Lin J,Fu Z J,Zhou Q Q,Chao Q L,Chen L,Yang Z B,Liu R Y. 2020. Metabolite signatures of diverse Camellia sinensis tea populations[J]. Nature Communications,11(1):5586. doi:10.1038/s41467-020-19441-1.
Zhang Q J,Li W,Li K,Nan H,Shi C,Zhang Y,Dai Z Y,Lin Y L,Yang X L,Tong Y,Zhang D,Lu C,Feng L Y,Wang C F,Liu X X,Huang J A,Jiang W K,Wang X H,Zhang X C,Eichler E E,Liu Z H,Gao L Z. 2020a. The chromosome-level reference genome of tea tree unveils recent bursts of non-autonomous LTR retrotransposons in driving genome size evolution[J]. Molecular Plant,13(7):935-938. doi:10.1016/j.molp.2020.04.009.
Zhang W Y,Zhang Y J,Qiu H J,Guo Y F,Wan H L,Zhang X L,Scossa F,Alseekh S,Zhang Q H,Wang P,Xu L,Schmidt M H,Jia X X,Li D L,Zhu A T,Guo F,Chen W,Ni D J,Usadel B,Fernie A R,Wen W W. 2020b. Genome assembly of wild tea tree DASZ reveals pedigree and selection history of tea varieties[J]. Nature Communications,11(1):3719. doi:10.1038/s41467-020-17498-6.
(責任编辑 陈 燕)
