无乳链球菌分子流行病学研究进展
2021-07-21刘静娴
赵 晶,刘静娴,刘 瑛
无乳链球菌(Streptococcus agalactiae),20世纪30年代由Rebecca Lancefield分离自患牛乳腺炎的奶牛而得名,因其具有兰氏B组抗原,又称被称为B族链球菌(Group BStreptococcus)。《伯杰氏细菌鉴定手册》将其描述为革兰染色阳性、触酶阴性的兼性厌氧菌,呈球形或卵圆形,直径小于2 µm。B族链球菌可在健康女性的生殖道和消化道中定植,青春期以后的女性检出率较高,在孕妇中可以达到17%~19%[1]。定植于孕产妇生殖道B族链球菌,可导致围产期婴儿感染,成为引起产褥期肺炎、脓毒血症和脑膜炎等侵袭性新生儿疾病的主要危险因素[2]。婴儿出生后7 d内发生的早发型感染(early onset disease,EOD)常伴发肺炎,1周后至3个月发生的迟发型感染(late onset disease,LOD)通常与脓毒血症和脑膜炎相关。大于3月龄的婴儿亦可被B族链球菌感染,称为超迟发型感染(long late onset disease,LLOD)。此外,B族链球菌还是产后感染和患有糖尿病、人类免疫缺陷病毒(HIV)感染、肿瘤等特定人群机会性感染的重要病原体,可引起菌血症、心内膜炎、皮肤和软组织感染及骨髓炎等[3]。自1938年首例致死性产后感染病例被报道,到上世纪70年代以来,B族链球菌已成为新生儿败血症和脑膜炎的主要病原体。在产前筛查联合预防性使用抗生素(intrapartum antibiotic prophylaxis,IAP)策略实施之前,它是造成发达国家新生儿死亡的头号感染因素。得益于IAP的广泛实施,近年EOD-B族链球菌感染率已显著下降,然而LOD患病率并没有明显变化,LOD-B族链球菌感染仍是新生儿发病和死亡的主要原因之一[4]。综上,了解全球B族链球菌在人群尤其是孕产妇体内定植和新生儿疾病中的分子流行病学特征以及分子流行病学分析方法十分重要。
1 B族链球菌分型和分子流行病学特征
1.1 荚膜血清型(capsular polysaccharide serotype,CPS)
B族链球菌菌体外包裹着一层荚膜多糖(capsular polysaccharide,CP),由CP决定的血清型称为CPS。根据B族链球菌CP的免疫反应性,目前已鉴定出10种血清型(Ⅰa、Ⅰb、Ⅱ、Ⅲ、Ⅳ、Ⅴ、Ⅵ、Ⅶ、Ⅷ、Ⅸ)和不可分型(nontypeable,NT)。CPS可以通过胶乳凝集法和多重PCR等分子生物学方法检测来鉴定。研究中常利用CPS将B族链球菌分离株分成血清学上相似的群体,以了解该菌在怀孕和非怀孕成人体内定植以及新生儿疾病中的流行情况。B族链球菌最常见的荚膜CPS包括Ⅰa、Ⅰb、Ⅱ、Ⅲ和Ⅴ,占全球血清型的98%[1]。然而,在世界各大洲之间,联合国定义的发达国家和非发达国家之间,不同血清型的流行率存在着差异(见表1)[5-26]。
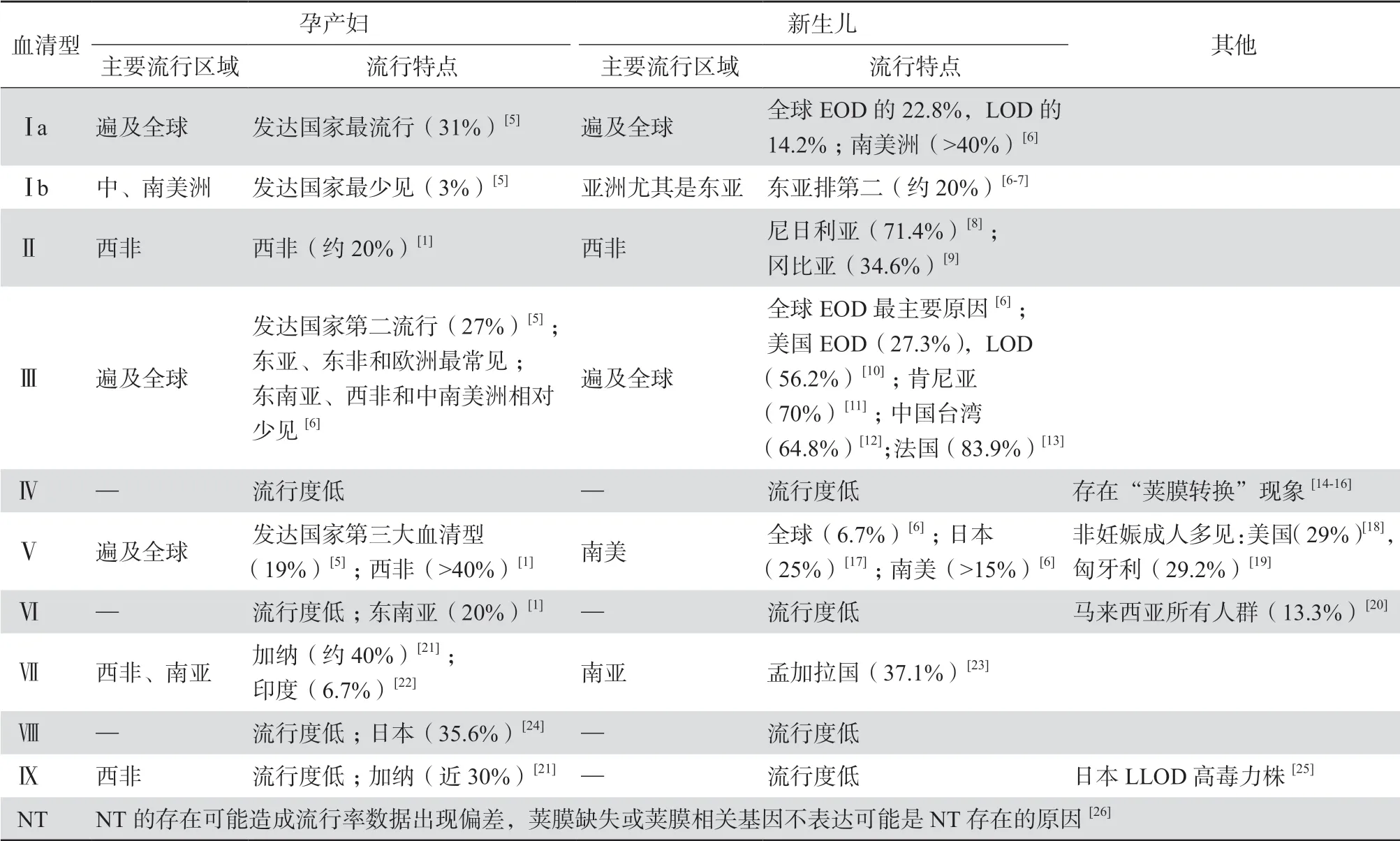
表1 无乳链球菌荚膜血清型全球流行分布及特点
1.2 多位点序列分型(MLST)
近年来,随着新的核酸检测技术出现,B族链球菌基因编码信息不断被破译,其分子流行病学研究得到了新的发展。目前发现,编码CPS的基因组,其包含的基因信息与特定的血清型并不完全一致。2003年,Jones等[27]利用7个已知的管家基因(adhP、atr、glcK、glnA、pheS、sdhA和tkt)建立了B族链球菌的MLST方法。随后Jolley等[28]创立了MLST数据库,该库现已收录4 000多个B族链球菌分离株信息。数据库显示目前全球主要流行的ST型包括ST-17(20.4%)、ST-1(13.6%)、ST-23(12.6%)、ST-19(7.9%)、ST-10(6.6%)[26]。
CPS与MLST序列类型(ST)有一定相关性,但并不是严格对应,具有相同ST的B族链球菌菌株可能有不同的血清型。数个ST可组成克隆复合体(clonal complexes,CC),同 一CC的ST成 员包含的上述7个管家基因中最多仅允许1个等位基因发生变异。CC的编号根据其祖系ST型或主要ST型确定。从人体分离的B族链球菌主要CC包括CC-17、CC-1、CC-23、CC-19、CC-10等[4]。从MLST全球数据库中可以得知各大洲B族链球菌分离株数据的分布差异,其中来自欧洲数据的占39.1%,非洲28.5%,亚洲13.4%,北美洲11.9%,南美洲0.1%,大洋洲数据缺少,而荷兰、肯尼亚、日本、美国、巴西分别提交了所属洲最多的数据[26]。需要指出的是,数据量占比并不代表其实际流行分布,主要与该地区的数据提交量有关。
从MLST数据库可以看到ST和CPS之间的某些关联性,每个最流行的ST及其所属的CC似乎都与某些特定的血清型相关,致病性亦有其相对独特的表现(见表2)。CC-17组的血清型同质性明显,除一小部分未明确的血清型外,所有分离株均关联CPS Ⅲ[26]。有研究指出,导致侵袭性新生儿感染的主要克隆株多属于Ⅲ型,且主要来自CC-17,占LOD-B族链球菌感染的80%以上[4]。前述我国台湾地区的研究中,ST-17占B族链球菌婴幼儿侵袭性疾病的56.6%,其中87.3%的ST-17为血清Ⅲ型[12]。CC-17以外的克隆株更多见于成人侵袭性感染。ST-12和CPS Ⅰb之间的相关性密切,而除了ST-17外其他所有ST中都发现有CPS Ⅴ菌株[29]。美国和加拿大的一项研究发现,CC-1组中的ST-1与CPS Ⅴ之间存在显著的相关性,而非妊娠期成人的侵袭性B族链球菌感染由Ⅴ型引起的占了相当大比重。该研究中的Ⅴ型分离株中ST-1占92%,非ST-1菌株最主要为ST-19。作者认为ST-1菌株的出现是20世纪90年代以来成人B族链球菌侵袭性感染的主要原因[30]。分离自人体的ST-1被认为可能与分离自牛的B族链球菌 ST-1型同源,全基因组测序确认了1992年的ST-1克隆株与20世纪70年代导致奶牛乳腺炎的瑞典菌株密切相关[4,30]。除此之外,部分B族链球菌克隆株还与定植有相关。前述Jones等[27]发现ST-19和部分ST-1与无症状定植显著相关,而ST-23在定植和侵袭性感染中均常见。来自加拿大的一项研究同样得出了类似的结论,认为ST-1和ST-23是孕妇中最常见的定植株[31]。而在非洲东北部,从孕妇阴道、直肠和新生儿体表分离的定植株主要克隆复合体为CC-10,占68.8%,其中ST-10最常见占37.5%[32]。综上所述,利用MLST分析B族链球菌的分子流行病学特征是近期的研究热点,这项技术的应用为我们提供了从血清型基因方面了解B族链球菌流行特点的新的角度。

表2 部分无乳链球菌MLST分型与荚膜血清型相关性
2 分子流行病学检测方法
2.1 脉冲场凝胶电泳(PFGE)
PFGE是另一种分析B族链球菌分子流行病学特征的分子生物学方法,在评估菌株遗传亲缘关系方面具有高区分度的特殊优势。有研究应用PFGE方法发现,血清Ⅰb、Ⅲ和Ⅴ型菌株具有同源性,而血清Ⅳ、Ⅰa和Ⅱ型菌株则不具有同源性[33]。这表明经典的CPS分析无法全面地揭示B族链球菌菌株之间遗传关系的远近。虽然使用PFGE可以观察到比传统血清学分型更为深入的菌株亲缘关系,但该技术也有其局限性。由于缺乏统一的命名分型系统,加之鉴定凝胶上的片段条带具有主观性,在报告条带大小时缺乏客观标准,PFGE分析很难在不同研究之间进行比较[26]。目前,该法多用于分析一项研究中来源不同的菌株及其传播方式之间的差异。例如,有项早期的研究观察到来自母体和其分娩儿的B族链球菌分离株具有相同的片段图谱模式,从而明确了该型菌株的母婴垂直传播[34]。从另一方面看,抛开其局限性,PFGE法揭示了B族链球菌菌株血清型内和血清型间存在的遗传变异,提供了从另一个角度研究B族链球菌分子流行病学特征的工具,促进了新的发展。
2.2 多位点可变数目串联重复序列分析(multiple locus variable-number tandem repeat analysis, MLVA)
MLVA原理与MLST相似,但该法利用的不是管家基因,而是VNTR(variable-number tandem-repeat)基因座。VNTR由遍布于整个细菌基因组不同基因位点的DNA的重复序列组成。不同菌株VNTR基因座的重复序列数量不尽相同,因此可以类似MLST,将几个VNTR基因座组合起来进行综合分析,从而产生不同菌株的VNTR特征性图谱。应用MLVA可以对多种细菌进行新的分型。Radtke等[35]研究了B族链球菌基因组5个最具多样性的基因座(SATR1、SATR2、SATR3、SATR4和SATR5),结果发现MLVA分型与CPS分型或MLST分型大体相符,但前者区分度更高。该研究中总共126株B族链球菌实验分离株具有70种不同的MLVA类型(MT),而只有36种MLST的ST型和19种CPS。同时该研究还显示,SATR2的重复序列与CPS Ⅸ型相对应,SATR3与ST-17密切相关,SATR5则与CPS Ⅴ/ST-1有一定相关性。SATR1基因座被认为是由成簇的规律间隔的短回文重复序列(clustered
regularly interspaced short palimdromic repeats,
CRISPR)组成的,其在MLVA分析中的作用尤为特殊和重要。
也有人仅使用6个VNTR基因座进行分析,包括上述Radtke等[35]研究的3个,同样显示了MLVA的98种 分 型 相 比MLST的51种 分 型 具有更大的区分力。该研究还发现,MLVA MT与MLST CC有相似之处,所有人体分离株CC-17均出现在MT-9群中。同时该研究还将MLST CC-23分成了CPSⅢ和CPS Ⅰa两群,再一次体现了MLVA的高区分度[36]。巴西一项研究对育龄非孕妇定植的B族链球菌利用MLVA进行基因分型,总共86株分离株中发现了15种MT[37]。另一项关于孕妇的研究中41株B族链球菌分离株中发现30株MT,同样显示了MLVA分型高度多样性的流行特征[38]。然而,与PFGE类似,因为没有统一命名标准的数据库,该分型方法同样很难在研究之间进行比较。但是MLVA分析显示出了高度区分力、高度多样性的B族链球菌分子流行病学特征,这一新技术的发展应用值得进一步关注。
2.3 CRISPR
CRISPR是原核生物基因组内的一段重复核苷酸序列,在生物进化史上,细菌为了将病毒、噬菌体的外来入侵基因清除,进化出了CRISPR-Cas系统。CRISPR和Cas(CRISPR相关蛋白)在细菌主动性防御和适应性免疫中起到重要作用。该系统保守而特殊的回文样基因排列及其变异,保留了细菌久远进化史上各个时期基因入侵的痕迹。上文提及的MLVA SATR1基因座即被认为是由CRISPR组成。在B族链球菌中,已经确定了两个CRISPR-Cas系统,包括普遍存在且有功能的II-A型系统和罕见且常不完整的I-C型系统,分别与CRIPSR locus 1 (CRISPR 1)和CRISPR 2相对应[39]。CRISPR1广泛存在于细菌基因组,使其成为追踪基因变异的一个非常重要的分子标记。利用这种方法,法国的一项研究能够在11年的时间里追踪监测100名女性的阴道B族链球菌定植演变情况[40]。另一项同样来自法国的研究则揭示了2群共存于孕妇体内的B族链球菌定植菌株群亲缘相近,可能来自同一祖先[41]。CRIPSR 1作为一种新的基因标志物,其记录了细菌随着时间演变形成的更为细致而又多样性的分子流行特征和流行趋势,成为了近年来的研究热点。
3 总结
本文综合阐述了目前全球无乳链球菌的分子流行病学特征,以及一些新的分子流行病学分析方法的研究应用。传统的CPS分型及其分布目前在全球研究得最为广泛和深入,其取得的大量数据,显示出了相对清晰的经典分子流行趋势。MLST是目前研究较多的技术领域,其描述的分子特征相对CPS更具多样性并有其自身的流行规律,提供了从基因层面了解B族链球菌流行特点的新角度。PFGE可用于分析分子特征相近菌株之间的遗传亲缘关系,在单项研究内了解血清型内和血清型间细微的遗传变异方面有其独特的优势。MLVA具有高区分度特点,其描述的细菌分子特征展现了B族链球菌更为复杂多样化的流行结构。CRISPR系统特有代代相传的基因入侵印迹,能让我们更为深入细致地了解菌株之间的遗传同源性。本文关于这些分子流行病学特征及研究进展的初步探讨,旨在为防治B族链球菌侵袭性疾病提供重要的流行病学资料,为血清型相关的疫苗研发和应用提供参考依据,为早期干预治疗母婴B族链球菌感染,减少母婴间垂直传播提供有价值的信息。全球各国学者对于B族链球菌分子流行病学的研究正在不断深入,前景值得期待。
