左乙拉西坦片仿制药与原研药溶出度一致性评价*
2021-07-15刘明鹭杨龙华朱双鹏
刘明鹭,李 震 ,杨龙华 ,朱双鹏,杨 钊
(1. 山东省青岛市食品药品检验研究院,山东 青岛 266000; 2. 浙江京新药业股份有限公司,浙江 绍兴 312500)
左乙拉西坦(LEV)为西坦类药物,属吡咯烷酮类,可视为 γ-氨基丁酸(GABA)的环状衍生物,是一种新型抗癫痫药物。左乙拉西坦片由比利时优时比制药公司[1]研发,商品名 KEPPRA(开浦兰),于 2000 年获美国食品药物管理局(FDA)批准,在美国和欧盟上市,并于2006 年11 月获国家食品药品监督管理局(SFDA)批准进口[1]。目前在全球超过66 个国家和地区上市,有超过100 万人的治疗记录,是目前美国癫痫治疗中心应用最多的抗癫痫药物[2]。仿制药与原研药的质量差异主要采用溶出度试验评价,通过在体外模拟口服固体制剂在胃肠道的溶出,以评价口服固体制剂的内在质量。而多种溶出介质下测定的溶出曲线可有效反映药物在体内的释放过程,本研究中比较并分析了左乙拉西坦片仿制药与原研药在4 种不同溶出介质中的溶出曲线,为评价仿制药质量和疗效的一致性提供参考[3-5]。现报道如下。
1 仪器与试药
1.1 仪器
Agilent 708DS 型溶出仪、Agilent 1260 型高效液相色谱仪(美国安捷伦公司);Sartorius Bt 125D 型天平(德国赛多利斯公司,精度为十万分之一);AS10200BT 型超声仪(美国赛默飞世尔科技公司,功率为300 W,频率为50 /60 Hz);Mettler MP200 型 pH 计(瑞士梅特勒公司);ZKT-18F 型真空脱气仪(天津天大天发科技公司)。
1.2 试药
左乙拉西坦对照品(A 公司,批号为R039L0,含量为99.9%);左乙拉西坦酸(杂质C)对照品(欧洲药品质量管理局,批号为4.0,含量为100.0%);左乙拉西坦片(仿制制剂,B 公司,批号分别为 A1802061,A1701202,A1701203,A1701204,规格为每片 0.25 g);左乙拉西坦片(原研制剂,商品名开浦兰,比利时优时比制药公司,批号为 195846,规格为每片 0.25 g);乙腈(色谱纯,美国Fisher 公司);纯化水(实验室自制);磷酸氢二钾、盐酸、无水乙酸钠、氢氧化钠(分析纯,国药集团化学试剂有限公司)。
2 方法与结果
2.1 色谱条件与系统适用性试验
色 谱 柱 :Agilent Eclipse XDB - C18柱 (150 mm ×4.6 mm,5 μm);流动相:0.2% 磷酸氢二钾溶液 - 乙腈(950 ∶50,V / V,用稀磷酸调 pH 值至 6.0);流速:1.0 mL / min;检测波长:205 nm;柱温:30 ℃ ;进样量:20 μL。分别取杂质C 对照品与左乙拉西坦对照品适量,加水 - 乙腈(950 ∶50,V / V)溶解,稀释,制成每 1 mL 各含0.1 mg 的溶液,作为系统适用性试验用溶液。理论板数按左乙拉西坦峰计应不低于3 000,杂质C 与左乙拉西坦峰之间的分离度应大于1.5。结果左乙拉西坦峰的理论板数为8 715,分离度为2.7。
2.2 溶出条件选择
首先采用《美国药典》(USP)收载的左乙拉西坦片的溶出条件对原研药进行测定[6],转速为 50 r/min,15 min时溶出量约为85%,观察溶出杯中崩解现象,原研药为溶蚀型,在50 r/min 条件下在杯底静水区沉积,薄膜包衣片明显崩解偏慢,推测在50 r/min 条件下可能存在过度区分。根据原国家食品药品监督管理总局发布的《普通口服固体制剂溶出度试验技术指导原则》[7]《普通口服固体制剂溶出曲线测定与比较指导原则》[8]《人体生物等效性试验豁免指导原则》[9],并参考文献[10 -11],溶出条件设定:溶出介质体积为500 mL,温度为(37±0.5)℃ ,转速为 75 r/min;4 种溶出介质(纯化水,经测定 pH 为 6.20),pH 1.0 盐酸溶液(取盐酸 9.0 mL,用水稀释至 1 000 mL,摇匀,即得),pH 4.5 醋酸盐缓冲液(取醋酸钠2.99 g,冰醋酸1.6 mL,加水溶解并稀释至1 000 mL,即得),pH 6.8 磷酸盐缓冲液(量取 0.2 mol/L磷酸二氢钾溶液250mL,0.2mol/L 氢氧化钠溶液112mL,混匀,加水稀释至1 000 mL,即得),上述溶出介质均采用脱气装置经42 ℃ 脱气1 h 后立即使用;取样时间为5,10,15,30,45 min。
2.3 溶液制备
取左乙拉西坦对照品25 mg,精密称定,置50 mL容量瓶中,分别用 pH 1.0 盐酸溶液、pH 4.5 醋酸盐缓冲液、pH 6.8 磷酸盐缓冲液和水4 种溶出介质溶解,并稀释至刻度,摇匀,制得每1 mL 中约含0.5 mg 的对照品溶液。取每批样品(批号分别为 195846,A1802061,A1701202,A1701203,A1701204)各 12 片,分别在 pH 1.0 盐酸溶液、pH 4.5 醋酸盐缓冲液、pH 6.8 磷酸盐缓冲液和水4 种溶出介质中进行试验,取溶出液,用 0.45 μm 聚醚砜微孔滤膜(天津市津腾实验设备有限公司)滤过,取续滤液,即得供试品溶液。
2.4 方法学考察
线性关系考察:取左乙拉西坦对照品50 mg,精密称定,置10 mL 容量瓶中,用水溶解,并稀释至刻度,摇匀,作为左乙拉西坦对照品贮备液,分别精密量取对照品贮备液 0.2,0.6,1.0,1.4,1.8 mL,置 10 mL 容量瓶中,用 pH 1.0 盐酸溶液、pH 4.5 醋酸盐缓冲液、pH 6.8磷酸盐缓冲液和水4 种溶出介质稀释至刻度,摇匀,按2.1 项下色谱条件进样测定。以左乙拉西坦对照品的质量浓度(X,mg/mL)为横坐标、峰面积(Y)为纵坐标绘制标准曲线,并计算回归方程。结果在上述4 种溶出介质中,左乙拉西坦对照品质量浓度在 0.10 ~0.90 mg/mL范围内与峰面积线性关系良好。详见表1。
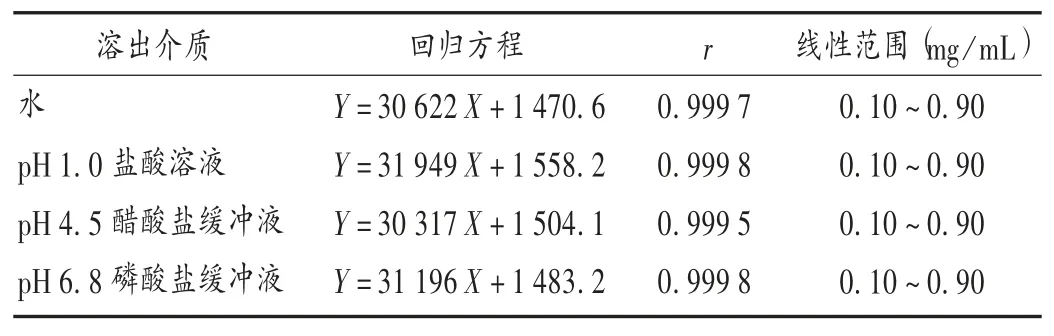
表1 线性关系考察结果(n =5)Tab.1 Results of linear relation test(n = 5)
精密度试验:取 2.3 项下对照品溶液,按 2.1 项下色谱条件重复进样测定6 次。结果水、pH 1.0 盐酸溶液、pH 4.5 醋酸盐缓冲液、pH 6.8 磷酸盐缓冲液 4 种溶出介质的 RSD 分别为 0.15% ,0.20% ,0.27% ,0.21%(n =6),表明仪器精密度良好。
稳定性试验:吸取2.3 项下4 种溶出介质45 min时的供试品溶液适量,分别于室温下放置0,2,4,8,16 h时,按2.1 项下色谱条件进样测定,结果左乙拉西坦的峰面积几乎无变化,水、pH 1.0 盐酸溶液、pH 4.5 醋酸盐缓冲液、pH 6.8 磷酸盐缓冲液的 RSD 分别为0.75% ,0.66% ,0.49%,0.70% (n =5),表明供试品溶液在室温条件下放置16 h 稳定性良好。另分别取4 种溶出介质在45 min 时的供试品溶液适量,置37 ℃水浴中保温,同法测定,结果的 RSD 分别为 0.83% ,1.06% ,0.79%,0.85%(n =5),表明供试品溶液在 37 ℃放置 16 h稳定性良好。
加样回收试验:取左乙拉西坦对照品适量,精密称定,置容量瓶中,按处方量加入辅料,分别用4 种溶出介质溶解,稀释,配制成相当于制剂中药物含量80%,100%,120%的浓度,混匀,制成低、中、高3 种浓度的溶液,各3 份,离心,按2.1 项下色谱条件进样测定,并计算加样回收率。结果见表2。
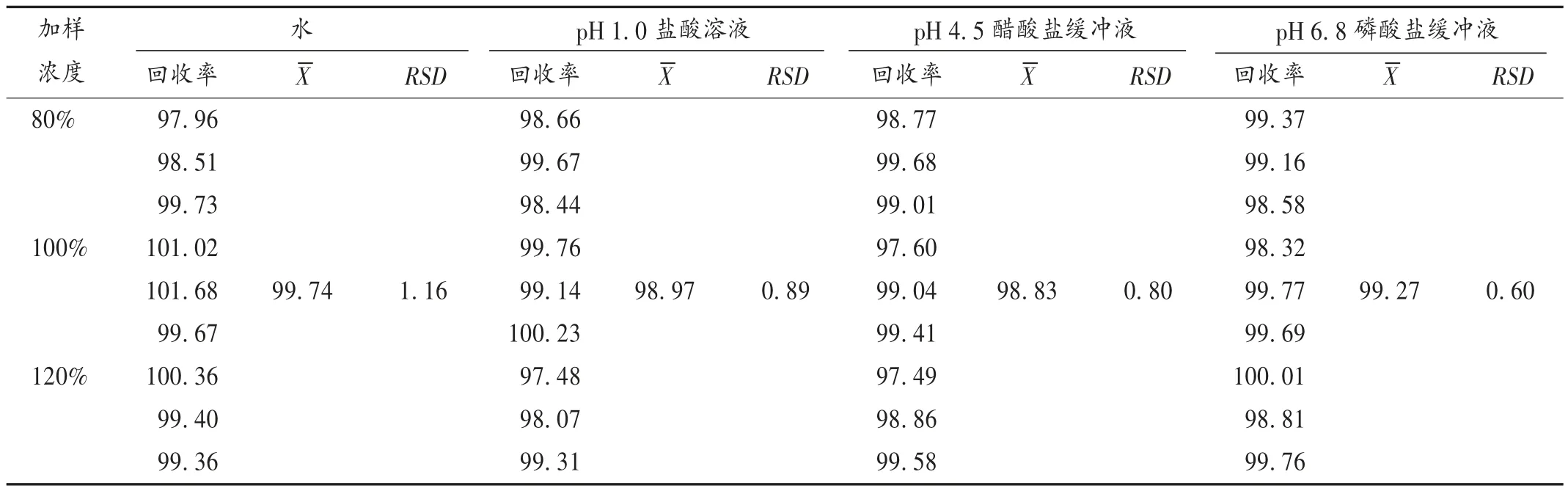
表2 4 种溶出介质中左乙拉西坦加样回收试验结果(%,n =9)Tab.2 Results of the recovery test of levetiracetam in four dissolution media(%,n = 9)
滤膜吸附试验:取左乙拉西坦原研药1 片,置500 mL容量瓶中,加500 mL 水,于 37 ℃水浴振摇 45 min,放冷至室温,摇匀,离心(转速为 5 000 r/min)10 min,0.45 μm 聚醚砜微孔滤膜滤过,分别弃去 1,2,3,4,5 mL 后,按 2.1 项下色谱条件进样测定,记录色谱图。结果弃去 1,2,3,4,5 mL 后的回收率分别为98.68% ,99.22% ,99.10% ,99.83% ,99.55% ,表明使用0.45 μm 聚醚砜微孔滤膜过滤对左乙拉西坦基本无吸附作用,不干扰测定结果。
管路吸附试验:在水的溶出曲线的最后1 个取样点分别进行手动取样和仪器自动取样,并进行含量测定,结果2 种溶出液测定峰面积基本一致,表明溶出仪管路对左乙拉西坦基本无吸附干扰。
2.5 溶出度测定
取1 批(批号为195846)左乙拉西坦片原研制剂与4 批(批号分别为 A1802061,A1701202,A1701203,A1701204)左乙拉西坦片仿制制剂(各12 片),按2015年版《中国药典(四部)》通则0931 第二法进行溶出度测定。分别取2.2 项下4 种溶出介质,依法操作,分别于5,10,15,30,45 min 时取溶出液 5 mL 后补充相同溶出介质,溶出液用0.45 μm 微孔滤膜滤过,取续滤液作为供试品溶液。取供试品溶液及2.3 项下对照品溶液,按2.1 项下色谱条件进样测定,并按外标法计算每片样品在各时间点的累积溶出度,绘制溶出曲线。详见图1。
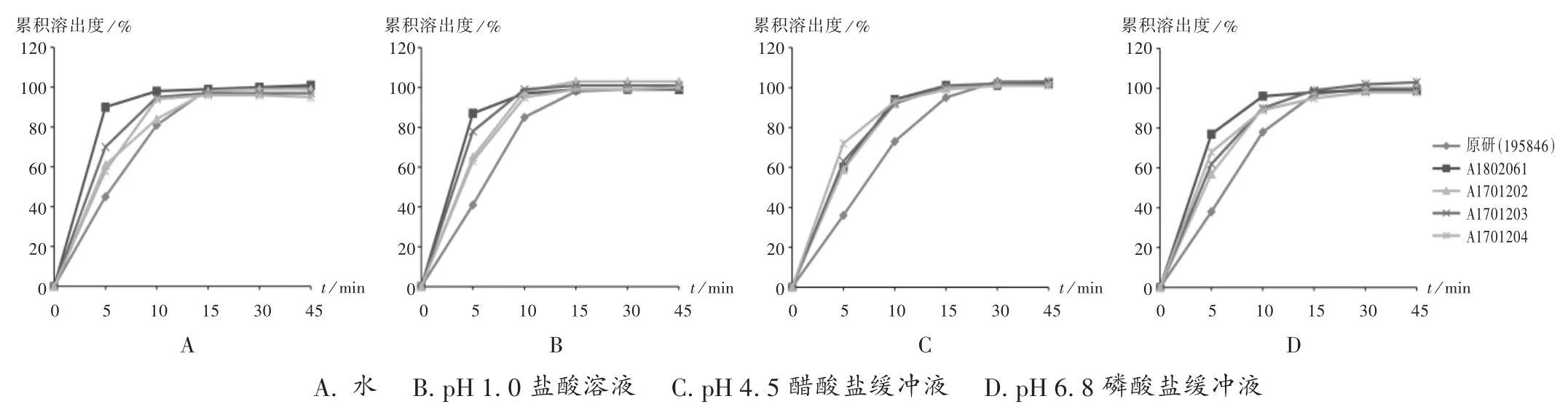
图1 仿制和原研左乙拉西坦片在4 种溶出介质中的溶出曲线(n =12)A. water B.pH 1.0 hydrochloric acid solution C.pH 4.5 acetate buffer solution D.pH 6.8 phosphate buffer solutionFig.1 Dissolution profiles of generic and original Levetiracetam Tablets in four dissolution media(n =12)
2.6 溶出一致性评价
由图1 可知,4 批仿制制剂与原研参比制剂在水、pH 1.0 盐酸溶液、pH 4.5 醋酸盐缓冲液、pH 6.8 磷酸盐缓冲液中15 min 时的溶出量均大于85%,可直接判定二者溶出行为相似,故不再计算 f2值[12]。
3 讨论
国务院于2012 年发布的《国务院关于印发国家药品安全“十二五”规划的通知(国安〔2012〕5 号》中明确提出,要推进仿制药质量一致性评价,全面提高仿制药质量[13]。测定在多种溶出介质中仿制药与参比制剂的溶出曲线,是多国评价口服固体制剂内在质量的重要手段,也广泛用于预测生物等效性[14]。本研究中选取4 批左乙拉西坦仿制药与1 批同规格原研药在4 种溶出介质中进行溶出度测定试验,并绘制溶出曲线,对比发现,仿制药与原研药在4 种溶出介质中的溶出曲线具有相似性,属于快速溶出,表明左乙拉西坦仿制药与原研药体外溶出行为一致,其质量一致性较好。
