杜氏肌营养不良症基因治疗新进展*
2021-07-13田国力王燕敏
纪 伟,田国力,王燕敏
上海市儿童医院,上海交通大学附属儿童医院 新生儿筛查中心,上海 200040
杜氏肌营养不良(Duchenne muscular dystrophy,DMD)和贝克肌营养不良(Becker muscular dystrophy,BMD),均由DMD 基因突变导致抗肌萎缩蛋白(dystrophin,Dys)缺失所致,是最常见的X 连锁隐性遗传性神经肌肉病。DMD 在存活男婴中的发病率为1/3000~5000[1]。在我国,每年大约有400~500 例DMD 患儿出生,总计DMD 患者高达7~8 万人,是患病人数最高的国家之一。患者最初通常在2~3 岁时出现临床症状,包括运动发育迟缓、步态异常、易跌倒等,肌肉组织的损伤导致患儿在8~12 岁时丧失行走能力并伴有多器官系统的受累,同时可出现认知、消化功能障碍以及心肌病等,大约在20 岁左右需要呼吸机辅助呼吸,20~30 岁时死于呼吸或循环衰竭[2]。BMD 是DMD 的轻度表现形式。
DMD 尚无有效治疗手段,经典的类固醇激素治疗结合多学科的综合评估和管理,可有效延长DMD 患者独立行走时间,提高患者的生存质量;但类固醇激素治疗伴有肥胖、矮小、骨质疏松等副作用,且不能改变疾病的最终结局。
随着基因技术发展,基因替代、外显子跳跃、基因组编辑、终止密码子通读等基因治疗逐步成为研究热点。而两种基于基因治疗的方法Ataluren 和Eteplirsen 已经分别被欧洲药物管理局(EMA)和美国食品药品监督管理局(FDA)批准。但是,这些药物只适用于一小部分患者,其他一些潜在的治疗方法都处于临床试验的晚期。本篇综述主要描述了基因治疗中最重要和最有前景的进展,并对其治疗优势及局限性进行分析。
1 基因替代治疗
基因替代治疗是在DMD 患者的基因组中插入外源的功能性DMD 基因,从而恢复DMD 患者骨骼肌、心肌细胞表达有功能的抗肌萎缩蛋白(如图1)。腺相关病毒(AAV)具有肌细胞取向性[3],可以作为DMD 基因载体。但AAV 非常小(4.5 kb),而DMD 基因是已知的人类最大基因(2.4 Mb)[4],因而利用病毒载体导入截短的、能表达部分Dys 的DMD 基因是基因替代治疗的一种选择。有研究发现,在DMD 基因突变小鼠(mdx 小鼠)模型中,局部注射缺少70%编码序列的微型Dys,可保护mdx 小鼠的四肢肌肉和心脏功能[5]。同样,在金毛犬肌营养不良症(golden retriever muscular dystrophy,GRMD)的犬模型中也实现了肌肉功能改善[3]。目前,针对AAV 介导的微型抗肌萎缩蛋白基因治疗正开展多项临床试验,也有研究通过双重腺相关病毒技术[6]和慢病毒载体[7]提高基因载体的容量,有望装载全长的DMD 基因。
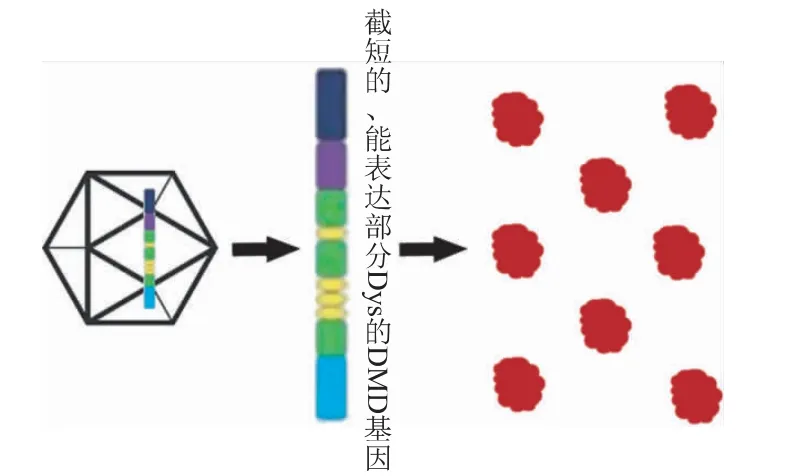
图1 基因替代治疗原理示意图
系统地将AAV 输送到骨骼肌、心肌进行基因治疗,对部分患者是可行的,但仍存在挑战。微型Dys 是否可改善肌肉功能尚仍不清楚。虽然微型Dys 在mdx 小鼠和GRMD 犬模型中实现了肌肉功能改善,但是mdx 小鼠肌肉质量优于人类DMD[8],并且不同犬之间肌肉萎缩程度存在很大差异。因此,根据动物模型的数据很难预测微型Dys 在人体中的作用[9]。
转基因表达的寿命有限。AAV 是一种游离体病毒,随着时间的推移,肌肉更新导致转基因逐渐丧失,从而使微型Dys 表达减少;但微型Dys 丢失将在基因导入多久后发生仍不清楚。GRMD 模型的一项研究显示,治疗3 个月后,45%的肌肉纤维中存在微型Dys,56 个月后,仅有5%的肌纤维中存在微型Dys[10]。转基因在体内究竟能表达多长时间仍不可知。
基因替代治疗不适用于体内含有抗AAV 抗体的患者,这类患者约占所有DMD 的50%[11],因对病毒衣壳有免疫反应,可能对微型Dys 本身产生体液或细胞免疫反应[12],故这类病人禁止接受AAV 基因疗法。按照治疗DMD 所需的病毒载体量,AAV 制造耗时且成本昂贵。使用小剂量治疗一名患者所需的病毒载体的生产周期通常需要1~2 月,而仅美国每年就有大约400 名DMD 患者出生。这就要求药企优化生产工艺,以便在更短时间内生产更多批量的病毒载体。
2 外显子跳跃治疗
外显子跳跃治疗是利用特异性反义寡核苷酸在DMD基因前信使RNA 剪接过程中,排除特定外显子以重建阅读框,将突变类型纠正为整码突变,这样大多数DMD 患者(见图2)在理论上可以产生截短的、但功能正常的Dys,是DMD治疗的又一策略[4](见图3)。约80%的DMD 突变可以通过跳跃1 到2 个特定的外显子纠正突变类型来恢复阅读框[13],其中,51 号外显子的跳跃可治疗约13%的DMD 患者[14]。
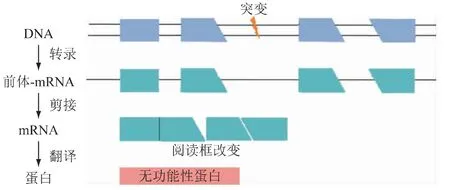
图2 DMD 突变示意图

图3 外显子跳跃治疗原理示意图
Eteplirsen 是专门为51 号外显子设计的一种反义寡核苷酸,2016 年9 月美国FDA 批准其用于DMD 治疗,这是首个通过FDA 批准的DMD 基因治疗药物。I、Ⅱ期临床试验已经证实了Eteplirsen 的安全性和耐受性,并在开放性非盲实验中进行的Ⅲ期临床试验,多数患者步行能力明显改善,对其进行肌肉活检发现Dys 和抗肌萎缩蛋白糖蛋白复合物的表达恢复[15]。经过3 年的持续治疗和随访显示,患者行走功能持续稳定,没有明显治疗相关副作用[16],证实Eteplirsen 可以改善患者运动,延缓呼吸功能的下降[17]。
Drisapersen 也是一种能够跳跃5l 号外显子的反义寡核苷酸,在近300 名DMD 患者中进行的一项非盲试验和三项安慰剂对照试验[18,19],结果显示,Drisapersen 治疗48 周后,治疗组中年龄为6~8 岁患儿比安慰剂组6 分钟步行距离(6MWT)有所延长[18];但是在全年龄段的所有DMD 患儿中,治疗组和安慰剂组的6MWT 无显著性差异[19]。另外,Drisapersen 治疗会有注射部位局部反应、一过性蛋白尿和发热等不良反应。最终Drisapersen 未获得FDA 和EMA 的批准。
同样的,外显子跳跃治疗也具有其局限性,例如转导不足、尤其是在心脏组织中,转导效率低下,组织摄入不稳定,需要频繁用药才能达到治疗效果。为进一步提高外显子跳跃治疗效率,对提高反义寡核苷酸的传递或吸收的研究正在进行。目前,仅在动物模型中测试了与反义寡核苷酸结合的肌导肽,它们使骨骼肌和心肌的外显子跳跃性水平提高了几倍[20]。富含精氨酸的肌导肽可导致组织非特异性摄取增加,这已经在临床前研究和临床试验中得到验证[21];但是富精氨酸的肌导肽即使在低于诱导外显子跳跃所需的剂量[22]也经常引起非人类灵长类动物和人类的肾毒性,因此限制了其临床的应用。
3 基因组编辑治疗
基因组编辑技术是利用细胞自身的修复机制——非同源末端链接(Non-homologous end joining,NHEJ)或同源重组(Homologous Recombination,HR)所致随机插入和/或缺失,在原基因座位修复突变基因。工程核酸酶可通过切割DNA 双链结构的特定序列,诱发修复位点断裂,从而达到原位基因编辑的目的(见图4)。
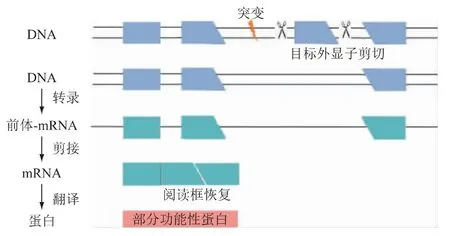
图4 基因组编辑原理示意图
成簇间隔短回文重复序列(clusteredregularly interspersed short palindromic repeats,CRISPR)上游基因所编码蛋白可在CRISPR 序列区诱导双链DNA 断裂,因此被命名为CRISPR 关联基因(CRISPR associated,Cas)。CRISPR/Cas 系统因其具有识别外源DNA 并靶向诱导双链DNA 断裂,以沉默外源基因表达的功能而被开发成一种高效的基因编辑工具。
从2016 年开始,CRISPR/Cas 系统逐渐在体外培养的肌卫星细胞、患者源性成肌细胞、人诱导多能干细胞源性肌肉细胞和mdx 小鼠模型中实现DMD 基因编辑,并可见Dys 水平恢复,部分骨骼肌和心肌肌力增强[23]。其中,人诱导多能干细胞源性肌肉细胞在目前研究中应用最广泛。有研究者[24]利用CRISPR/Cas9 系统,通过NHEJ 修复45—55 外显子之间的长达725kb 的突变片段,是目前CRISPR/Cas9 介导的基于NHEJ 修复的最大片段的DMD 缺失。另外,小鼠体内研究显示,经基因组编辑治疗获得的多能干细胞分化形成的心肌与骨骼肌,可恢复正常Dys 水平。而采用患者自身诱导性多能干细胞在体外进行基因编辑修正后输入患者体内,尽可能避免异体移植带来的免疫排斥反应,这是未来研究的新方向。
2018 年,Amoasii L 等[25]首次成功利用CRISPR/Cas9 技术对50 号外显子缺失的DMD 犬模型完成外显子跳跃治疗。次年,Min YL 等[26]通过向44 号外显子缺失的小鼠模型中注射重组AAV 包装的Cas9 核酸酶和单向导RNA,可以恢复抗肌营养不良蛋白表达以及肌肉收缩能力的改善。这些研究表明,CRISPR-Cas 系统可以用于纠正导致DMD 的多种遗传突变,并为永久纠正DMD 提供了潜在的基因治疗前景。
然而,目前基因编辑治疗仍处于体外细胞和动物实验阶段,对于应用于人体的安全性和有效性还需进一步临床试验证实。基因组编辑是在DNA 水平上进行的,因此被编辑基因产生的所有RNA 转录本来都是自身产生的,理论上这就排除了重复治疗的需要;但基因编辑治疗不能完全预防与DMD 相关的肌肉损伤,一旦发生大规模的肌肉损伤,多核纤维中前期已接受基因组编辑过的细胞核就会丢失。因此,患者仍有可能随着时间的推移,部分Dys 水平逐渐下降。DMD基因组编辑治疗的长期有效性,还需进行长期的研究。
基因组编辑治疗也同样面临着机体对AAV 的免疫抵抗以及对AAV 载体的大量需求。此外,基因编辑方法也面临着独特的挑战,比如,已知Cas9 可以在目标位点以外的地方诱导DNA 断裂[27],产生脱靶效应,导致DNA 的永久改变。
由于DMD 基因突变类型的多样性,单一设计的CRISPR/Cas9 基因编辑方法不能完全治疗所有类型DMD 患者,未来还需研究发现更加特异、高效的内切酶工具,提高基因编辑的安全性、有效性,加速最终治疗DMD 患者的进程。
4 终止密码子通读治疗
无义突变是由于某单一碱基突变使氨基酸密码子转变为终止密码子的点突变,提早出现的终止密码子引起mRNA退变,提前终止蛋白质翻译,产生截短的、无功能的蛋白质。终止密码子通读治疗又称为无义突变通读治疗,一些化合物通过与核糖体的结合,阻止终止密码子的信号识别,从而跳过这个错误的终止密码子,诱导提早出现的终止密码通读,从而继续翻译Dys(见图5)。该方法适用于所有无义突变DMD 患者,其占总患者人群的10%~15%[28]。

图5 终止密码子通读治疗原理
研究发现,在mdx 小鼠中,庆大霉素可诱导Dys 的高表达,增加肌肉的收缩能力[29];但庆大霉素的耳毒性和肾毒性限制了在DMD 治疗中的应用。高通量筛选已经确定了Ataluren(PTC124,商品名Tranlsarna)为一种蛋白修复药物,能够降低核糖体对过早终止密码子的敏感性,具有潜在的终止密码子读取能力,造成所谓“终止密码子通读”[30],且具有比庆大霉素更好的安全性。
一项治疗前后的比较研究发现,Ataluren 可使Dys 表达增加11%[31]。Bushby K 等[32]进行了一项随机双盲多中心试验,174 例5~20 岁的无义突变DMD 的男性患儿,接受了为期48 周的Ataluren 治疗,治疗中无严重不良事件报道,Ataluren 具有良好的安全性与耐受性,40 mg·(kg·d)-1剂量治疗组比对照组6MWT 增加了31.3 m。荟萃分析结果显示,Ataluren 对6MWT 为300~400 m 的患者治疗效果显著[33],且可以维持肌肉功能。2014 年EMA 对年龄≥5 岁的无义突变患者予以有条件批准使用Ataluren 治疗。Ataluren 的主要缺点是只适用于具有无义突变的个体(约13%的DMD 患者[34]),并且与其他治疗罕见疾病的药物一样,价格昂贵。由于代理商的市场和价格在每个国家都是单独协商的,因此Ataluren目前只在欧洲个别国家应用。
5 小 结
DMD 的治疗在近十几年中发展迅速,特别是在基因治疗领域,包括治疗方法的多样化及基因载体的不断改进,整体前景令人期待。目前已有两种药物获得批准,另有数十种药物正在进行临床试验,众多的治疗方法对DMD 小鼠和GRMD 的犬模型中都已经展现出较好的治疗效果;但应用于临床仍需克服许多障碍,有待于进一步优化和更多的研究支持。基因替代疗法、外显子跳跃技术、基因组定向编辑以及终止密码子通读治疗适用于不同类型的DMD 患者,都有着各自的优势和不足之处(如表1 所示),相信曾经无药可治的DMD 在不久的未来会在研究者们共同的努力下最终被攻克。

表1 4 种基因疗法归纳总结
