HPLC-QAMS 法同时测定芪冬颐心颗粒中7 种成分的含量*
2021-07-13乔春凤陈海燕
刘 华,乔春凤,陈海燕
1 郑州颐和医院 药学部,郑州 450047;2 广西卫生职业技术学院 药学系,南宁 530023
芪冬颐心颗粒由黄芪、麦冬、茯苓、人参、地黄、烫龟板、淫羊藿、煅紫石英、桂枝、金银花、丹参、郁金和炒枳壳等13 味中药材加工而成,现收载于《中国药典(2015 年版一部)》,主要用于气阴两虚所致的心悸、胸闷、胸痛、气短乏力、失眠多梦、自汗、盗汗、心烦等症候的治疗,亦可用于病毒性心肌炎、冠心病心绞痛见上述症候的治疗[1]。中成药复方制剂是多成分的集合体,通过多个成分间相互协同作用达到临床的治疗效果,近年来,多指标成分质量控制模式已逐步应用于中药及其制剂中。芪冬颐心颗粒现行单成分质量控制方法[1]难以全面评价其产品质量,尚未见对芪冬颐心颗粒所含成分进行定量研究的文献报道,因此选取淫羊藿苷为内参物,采用高效液相色谱一测多评法对芪冬颐心颗粒中主要成分毛蕊异黄酮葡萄糖苷、芒柄花素、朝藿定A、淫羊藿苷、宝藿苷I、去氢土莫酸和茯苓酸的含量进行同时测定,建立芪冬颐心颗粒多指标成分质量控制模式,为全面有效地控制产品整体质量提供实验数据支持。
1 仪器与药品
Agilent 1290 型高效液相色谱仪(美国Agilent 公司);Waters e2695 型高效液相色谱仪(美国Waters 公司);XP105 型电子天平(瑞士Mettler Toledo 公司)。
芪冬颐心颗粒(规格:每袋装5 g,批号:190201、190302、190304)来源于沈阳东新药业有限公司;毛蕊异黄酮葡萄糖苷对照品(批号:111920-201606,纯度:97.6%)和宝藿苷I 对照品(批号:111852-201603,纯度:99.9%)均来源于中国食品药品检定研究院;芒柄花素对照品(批号:PRF8091225,纯度:99.9%)来源于成都普瑞法科技开发有限公司;去氢土莫酸对照品(批号:CFS201802,含量:98.0%)来源于武汉天植生物技术有限公司;朝藿定A 对照品(批号:18050224,纯度:96.5%)、淫羊藿苷对照品(批号:18052121,纯度:98.2%)、茯苓酸对照品(批号:18053121,纯度:99.5%)均来源于上海同田生物技术股份有限公司。乙腈为色谱纯,其余试剂均为分析纯。
2 方法与结果
2.1 对照品贮备液和混合对照品溶液的制备
精密称取毛蕊异黄酮葡萄糖苷、芒柄花素、朝藿定A、淫羊藿苷、宝藿苷I、去氢土莫酸和茯苓酸对照品适量,用甲醇制成浓度分别为0.316、0.718、0.454、0.982、0.596、0.118、0.172 mg·mL-1的混合对照品贮备液。精密吸取混合对照品贮备液2.5 mL,用甲醇定容至50 mL,制成浓度分别为15.8、35.9、22.7、49.1、29.8、5.9、8.6 μg·mL-1的混合对照品溶液。
2.2 供试品溶液和阴性供试品溶液的制备
取芪冬颐心颗粒适量,混匀研细,取粉末1.0 g,精密称定,加入25 mL 甲醇,称重,超声提取30 min,放冷后称重,用甲醇补重后过滤,制成芪冬颐心颗粒供试品溶液。按照《中国药典(2015 年版一部)》芪冬颐心颗粒“质量标准”项下的处方工艺,分别制备缺黄芪、缺淫羊藿或缺茯苓的阴性样品,再按上述方法制备成3 种阴性样品溶液。
2.3 色谱条件
色谱柱:Agilent TC-C18柱(250 mm×4.6 mm,5 μm),柱温:30 ℃;检测波长:254 nm(0~25.0 min检测毛蕊异黄酮葡萄糖苷和芒柄花素)[2]、270 nm(25.0~42.0 min 检测朝藿定A、淫羊藿苷和宝藿苷I)[3,4]和210 nm(42.0~60.0 min 检测去氢土莫酸和茯苓酸)[5,6];流动相:乙腈(A)-0.1%磷酸水溶液(B),梯度洗脱(0~14.0 min,16.0%A;14.0~25.0 min,16.0%A→24.0%A;25.0~42.0 min,24.0%A→45.0%A;42.0~54.0 min,45.0%A →82.0%A;54.0~60.0 min,82.0%A→16.0%A);流速:1.0 mL·min-1;进样量:10 μL。
2.4 专属性试验
精密吸取“2.1”项下制备的混合对照品溶液和“2.2”项下各溶液依法进样测定,结果在供试品溶液色谱图中毛蕊异黄酮葡萄糖苷、芒柄花素、朝藿定A、淫羊藿苷、宝藿苷I、去氢土莫酸和茯苓酸峰形对称,与相邻色谱峰的分离度均>1.5,理论塔板数按各目标成分色谱峰计均≥4000,上述成分的含量测定无干扰,色谱图见图1。

图1 混合对照品(A)、芪冬颐心颗粒(B)、黄芪阴性样品(C)、淫羊藿阴性样品(D)和茯苓阴性样品(E)的HPLC 色谱图谱
2.5 标准曲线的制定
精密吸取 “2.1” 项下混合对照品贮备液0.1、0.5、1.0、1.5、2.0、2.5 mL,分别用甲醇定容至20 mL,制成线性考察混合对照品溶液,依法进样测定,记录毛蕊异黄酮葡萄糖苷、芒柄花素、朝藿定A、淫羊藿苷、宝藿苷I、去氢土莫酸、茯苓酸的峰面积,以质量浓度(c,μg·mL-1)为横坐标,峰面积(A)为纵坐标进行线性回归,结果见表1。

表1 7 种成分的回归方程、线性范围和相关系数
2.6 进样精密度、重复性和稳定性试验
取“2.1”项下混合对照品溶液重复进样6 次,测得7 种成分峰面积的相对标准偏差(RSD) 分别为1.02%、0.62%、0.85%、0.59%、0.68%、1.23%、1.11%。
取同一批号芪冬颐心颗粒适量(批号:190201),按“2.2”项下方法平行制备6 份供试品溶液,进样测定各成分的峰面积,测得7 种成分含量的RSD 分别为1.65%、1.16%、1.29%、0.97%、1.02%、1.81%、1.78%。
取芪冬颐心颗粒(批号:190201)的同一份供试品溶液,于0、2、4、6、12、18 h 进样,检测7 种成分的峰面积,结果芪冬颐心颗粒供试品溶液18 h 内稳定,7 种成分峰面积的RSD 分别为0.99%、0.64%、0.86%、0.57%、0.71%、1.19%、1.08%。
2.7 回收率试验
取7 种成分含量已知的同一批次芪冬颐心颗粒(批号:190201)适量,除去内包装,混匀研细,每份0.5g,取9 份,精密称定,随机分成3 组,每组3 份,按《中国药典(2015 年版四部)》要求,精密加入混合对照品溶液(毛蕊异黄酮葡萄糖苷0.113mg·mL-1、芒柄花素0.241mg·mL-1、朝藿定A0.169mg·mL-1、淫羊藿苷0.354mg·mL-1、宝藿苷I0.213mg·mL-1、去氢土莫酸0.041 mg·mL-1、茯苓酸0.059mg·mL-1)1.0、2.0、3.0mL 各一组,使各成分对照品加入量约为样品含有量的50%、100%、150%,再按照“2.2”项下方法制备加样供试品溶液,依法进样测定毛蕊异黄酮葡萄糖苷、芒柄花素、朝藿定A、淫羊藿苷、宝藿苷I、去氢土莫酸、茯苓酸的峰面积,采用外标法计算7 种成分的加样回收率,其平均加样回收率及相对标准偏差(RSD)分别为97.44%(0.93%)、99.60%(0.86%)、99.21%(1.36%)、100.03%(0.75% )、98.77%(1.08%)、97.64%(1.22%)、96.86%(1.07%)。
2.8 相对校正因子(RCF)的测定
依据“2.5”项下6 种线性关系(见表1),精密吸取混合对照品溶液适量,依法进样测定,记录毛蕊异黄酮葡萄糖苷、芒柄花素、朝藿定A、淫羊藿苷、宝藿苷I、去氢土莫酸、茯苓酸的峰面积,以淫羊藿苷为内参物,按照计算公式:RCF=fS/fR=
(式中c 代表质量浓度,A 代表峰面积,S 代表内参物,R代表其他待测组分)分别计算6 种成分的RCF,结果见表2。

表2 6 种成分的相对校正因子
2.9 RCF 耐用性考察
精密吸取“2.1”项下混合对照品溶液,依法进样测定毛蕊异黄酮葡萄糖苷、芒柄花素、朝藿定A、淫羊藿苷、宝藿苷I、去氢土莫酸、茯苓酸的峰面积,分别考察高效液相色谱仪(Agilent 1290 型、Waters e2695 型)、色谱柱(Agilent TC-C18柱、Phenomenex C18柱、Waters XTerra MS C18柱)、柱温(28 ℃、29 ℃、30 ℃、31 ℃、32 ℃)和流速(0.8、0.9、1.0、1.1、1.2 mL·min-1)对RCF 的影响。结果显示在不同仪器、色谱柱条件下,毛蕊异黄酮葡萄糖苷、芒柄花素、朝藿定A、宝藿苷I、去氢土莫酸、茯苓酸的平均对相对校正因子分别为1.3037、0.9395、1.6854、1.0760、2.0079、1.5491,RSD 分别为0.48%、0.69%、1.03%、0.86%、0.49%、0.67%;不同柱温条件下,各成分的平均相对校正因子分别为1.3039、0.9453、1.6918、1.0764、2.0154、1.5515,RSD 分别为0.46%、0.95%、1.19%、1.39%、0.32%、0.63%;不同流速条件下,各成分的平均相对校正因子分别为1.3019、0.9409、1.6878、1.0726、2.0161、1.5469,RSD 分别为0.35%、0.82%、1.01%、1.34%、0.49%、0.83%。结果表明,所建立的RCF 耐用性良好。
2.10 待测组分色谱峰的定位
精密吸取“2.1”项下混合对照品溶液,依法进样测定,记录毛蕊异黄酮葡萄糖苷、芒柄花素、朝藿定A、淫羊藿苷、宝藿苷I、去氢土莫酸、茯苓酸色谱峰的保留时间,以内参物淫羊藿苷色谱峰为基准峰,在不同品牌高效液相色谱仪(Agilent 1290 型、Waters e2695 型)和不同品牌色谱柱(Agilent TC-C18柱、Phenomenex C18柱、Waters XTerra MS C18柱)条件下,采用相对保留时间值法对色谱峰进行定位,结果毛蕊异黄酮葡萄糖苷、芒柄花素、朝藿定A、宝藿苷I、去氢土莫酸、茯苓酸的平均相对保留时间分别为0.5118、0.6468、0.9270、1.1335、1.4188、1.5605,RSD分别为1.51%、0.98%、0.69%、0.80%、0.36%、0.83%。
2.11 样品含量测定
取3 个不同批号的芪冬颐心颗粒,按“2.2”项下方法制备供试品溶液,每个批号平行3 份,依法进样测定毛蕊异黄酮葡萄糖苷、芒柄花素、朝藿定A、淫羊藿苷、宝藿苷I、去氢土莫酸、茯苓酸的峰面积,采用外标法和一测多评法分别计算上述7 种成分的含量。见表3。
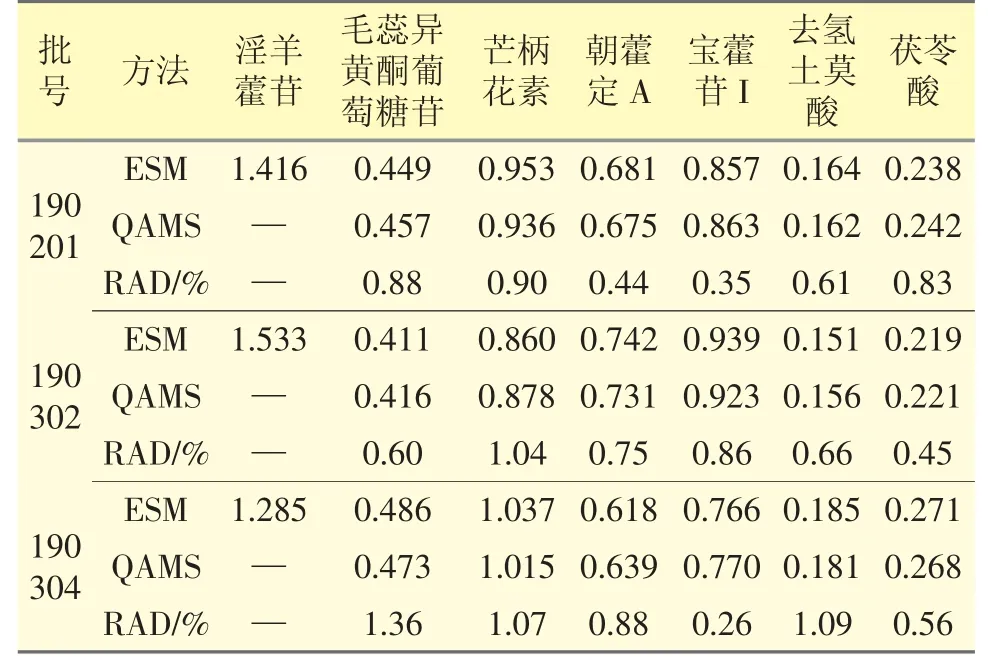
表3 7 种成分含量测定结果(mg·g-1,n=3)
3 讨论
3.1 流动相的筛选
本实验在芪冬颐心颗粒中测定目标7 种成分的色谱峰的峰形及分离效果,同时兼顾杂质成分的干扰情况,参考相关文献,依次对比考察了乙腈-水[3,4]、乙腈-0.1%磷酸水溶液[5,6]、乙腈-0.1%甲酸水溶液[2]流动相系统,结果发现,乙腈-0.1%磷酸水溶液系统目标成分分离效果较好,色谱图基线较为平稳且杂质干扰较小,通过对流动相比例不断摸索优化,最终确定采用乙腈-0.1%磷酸水溶液为流动相。按照“2.3” 项下流动相比例梯度洗脱可用于芪冬颐心颗粒中7 种成分含量的同时测定。
3.2 供试品制备方法的确定
本实验在芪冬颐心颗粒中测定目标7 种成分综合提取率,同时兼顾分析检测时间及杂质干扰程度,依次对比考察了提取溶剂(甲醇[5,6]、稀乙醇[3,4])和提取方式(超声提取[5,6]、加热回流提取),结果发现,甲醇超声提取效果最佳,还对提取时间进行摸索优化,最终确定采用甲醇超声提取30 min 制备芪冬颐心颗粒供试品溶液。在此条件下,7 种成分能够提取完全,在60 min内能够完成7 种成分的检测,杂质干扰较小。
4 小 结
本实验运用HPLC-QAMS 法对芪冬颐心颗粒中7 种成分含量进行了同时测定,建立其多指标成分质量控制模式。与外标法比较,各成分含量测定结果无显著差异(见表3)。本方法操作便捷、结果准确,为全面评价芪冬颐心颗粒整体内在质量、提升该颗粒剂质量标准提供了参考依据,以确保临床用药的稳定性和治疗效果的一致性。
